Free Energy Perturbation (FEP) Using QUELO¶
Overview¶
QUELO provides the user with the ability to run relative free energy calculations, using the free energy perturbation (FEP) approach. QUELO’s key features include:
Ability to run FEP using a next-generation molecular mechanics (MM) force field
Powerful artificial intelligence (AI)-based parametrization of the ligand, seamlessly integrated with the receptor and solvent force field terms. This is trained to mimic the behavior of quantum mechanics
Ability to run FEP using a combined quantum mechanics (QM)/molecular mechanics (QM/MM) force field
Both the bound ligand and interacting protein residues in the binding region can be treated using QM
Hundreds of atoms can be treated at the QM level without any sacrifices in sampling or the FEP approach
Fully automated: A simple-to-use interface replaces complicated setup and workflow for the user
From the user standpoint, performing FEP using a QM/MM force field is nearly identical to performing FEP using a traditional classical MM force field. The only difference is that in the QM/MM case, the user needs to define the region of the system that will be treated using QM. This is simply done using options provided through the interface.
The FEP calculation is fully automated to make these calculations simple to carry out. Among the tasks that are automatically handled on the backend when a calculation workflow is started are:
Parametrization for MM regions
Partitioning between the QM and MM regions (for QM/MM simulations)
Creation of a periodic solvation lattice
Superposition of ligands to a bound reference ligand
Creation and execution of an optimized set of calculations necessary to determine the relative free energies for the full input set
Analysis and compilation of results
Parallelized distribution of tasks
Below, the QUELO interface is described in detail.
The FEP Task List¶
When you enter the platform, you will be presented with the FEP Task List, a list of calculations (tasks) that you have previously set up and/or run, as well as a dialog to create a new task. Clicking on a task will bring you to the setup/results page for that task.
For more details on the FEP Task List, see the section The Task List.
Expert Mode¶
A number of options (described below) are only shown in Expert Mode. The options shown in (default) Standard Mode are sufficient for most users to run a reliable simulation. If you need access to Expert Mode, that is accomplished via a toggle in the User Settings panel.
For more details on enabling Expert Mode, see the section Settings and Expert Mode.
File Input Specification¶
When you click on a task with Staged status in the FEP Task table, you enter the setup dialogs for that task. At the top of the setup, you will need to specify the input files that are used to run the calculation. These files provide structural information on the protein receptor, as well as a set of ligands bound to the receptor for which free energy calculations will be carried out.
Note that after you submit a calculation to run, in some cases you may decide you wish to add additional ligands to the compute set. This is possible, and the process for this is described in its own section (Adding Additional Ligands to a Computed Set) later in this document.

A FEP calculation will calculate the relative free energies of binding of a set of ligands to a common protein receptor. Limitations of a standard FEP implementation require that both ligands be broadly similar to one another.
To run a calculation, the following components must be defined by the user, using the Browse/Upload dialogs in this section of the panel.
Errors, Warnings, and Deleting an Input Structure¶
After an input file is uploaded, the contents of that file will be summarized below the Upload button. The summary will include the name, the file name, and an Information field, which will be populated with an informative message when there is a warning or error related to the upload. In the case of an error/warning, in addition to a message in the Information field, the Name and file name will be colored red (rather than black) so that it is readily apparent when there is an issue with the input. There is also a red X at the right of each summary line, which allows you to delete the uploaded molecule, if desired.
Protein Receptor¶
This is the common protein receptor to which all the ligands that will be scored are binding. It is assumed that all ligands bind to the same receptor site, and that a single input conformation of the receptor binding site is suitable for all ligands.
The input format of the receptor is PDB. Care should be taken to ensure that the input PDB file is suitable for the calculation. See the chapter Preparing a PDB File for Use with QUELO for detailed information on how this file must be processed before using it with QUELO. QUELO expects a structurally complete receptor, formatted in a consistent way. Preparing the structure can be performed either by the user with third-party tools, or with our tool for PDB preparation, ProtClean: The Protein Preparation Tool. If the structure is not prepared with ProtClean, it should at least be run through ProtClean in its Format Only mode to ensure proper formatting before being uploaded to QUELO.
Issues that will need to be addressed before import include
All atoms must be present with a correct element entry, including hydrogen
Removal of symmetry replicates from the unit cell
Addition of missing loops near the binding region
Ensuring that the residue and atom naming convention is consistent with that expected by QUELO
Possible removal of unnecessary cofactor molecules, and the proper specification of their topology
Possible identification of structurally-important waters and removal of other waters
A properly prepared input protein receptor file is critical to obtaining high-quality results, and the user should read the PDB preparation and ProtClean chapters carefully.
Reference Ligand¶
The user must ensure that at least one ligand is pre-oriented properly and with high confidence in the binding site. For example, the user may obtain this ligand from a crystal structure of the complex or from modeling. This ligand must be supplied with 3D coordinates (SDF or MOL2), and it will be used as the reference binding conformation for any other ligands that need to be reoriented. The reference ligand must include all atoms, including hydrogens.
Whether you use an SDF or MOL2 format reference ligand input structure, the structure must include a full description of topology information. This requirement ensures that the program properly resolves any structural ambiguities. If you wish to use a bound ligand in a protein/ligand complex structure as your reference ligand, you will need to create a file that contains only the ligand structure and upload it here, separately, to use it as the reference ligand, and you will need to convert the format of the ligand to SDF or MOL2.
The user must specify a name for the reference ligand that is being uploaded in the appropriate box (to the left of the Upload button). If this name is not supplied, the Upload button will not be activated.
All Ligands (Excluding Reference)¶
The user can specify any number of additional ligands for which FEP calculations will be performed. These ligands are provided as 3D structures in either SDF or MOL2 format and should be located in the same binding pocket as the reference.
You can supply more than one input file of input molecules. After one file is imported, clicking Browse and then Upload for additional files will append the molecules in the subsequent files to the bottom of the list.
For each molecule in the input, a Maximum Common Substructure (MCS) alignment with the reference ligand will be carried out to align the 3D structure with the reference ligand.
After the input file for this section is uploaded and parsed, the names and canonicalized SMILES representations for all the ligands will be shown in a table below the input dialog line. For each input structure, the name is taken from the input file (if provided) or is otherwise the LNNN name auto-generated by the platform (L001, L002, … for each ligand without a user-provided name). If you click on the red X at the end of any input ligand line, that ligand will be removed from the input (a confirmatory pop-up will appear when you click on the X to ensure you don’t inadvertently delete a structure). Additionally, you will find a red X in a bolder font at the top of the column of red X buttons. If you wish to delete all the added ligands in this section at once, click on this bolder X.
Note that no two ligands can be identical. If identical ligands are found in the input, these will be annotated as errors in the Information field and shown in red.
You must remove (using the red X) all problematic ligands before you can proceed with the calculation.
Expert Mode options for this section are highlighted in the sample figure by red boxes. These options are only presented when you have enabled Expert Mode. They are described below:
Solvent already generated (Expert Mode)¶
Default: OFF
Turn on this option if you wish to supply a receptor input with a pre-generated solvent box. By default, QUELO will automatically generate the complete solvent box around the target. Crystal water molecules are retained unless they fall outside this box. If this option is switched on, QUELO will instead assume the solvent box is already generated and accept what is provided in the uploaded protein receptor. This gives the user full control over the size, structure, and composition of the solvent box. This option needs to be switched on to perform calculations on a membrane-bound system. The size of the solvent box must be specified at the top of the receptor PDB file in a “CRYST1” line. The solvent box must be cubic. The line should be consistent with the example below. The first three numbers are the dimensions of the solvent box (in angstrom) and the second three are angles (which should be 90.00), defining the box as cubic.
CRYST1 102.406 102.406 82.314 90.00 90.00 90.00
Ligand alignment (Expert Mode)¶
Every additional ligand input needs to be aligned on the reference ligand before a FEP calculation can proceed. The simplest option (2D alignment) for alignment uses just the 2D topology of the maximum common substructure (MCS). The default option (3D alignment) is a generally better approach using MCS metrics based on 3D structural information rather than just the 2D topology.
A third option (perfectly prealigned) is to upload ligands that are already perfectly aligned and to use that alignment to define the perturbation map and FEP single/dual regions. Unlike the 3D and 2D alignment approaches, this alignment is based on how the coordinates of the atoms line up as provided, with no attempt to gently relax the ligand structures and maximize alignment. Atoms will only be considered aligned if they are topologically identical (with the same element and hybridization) and within 0.1 angstrom of each other. Use this option carefully, as it can result in very small single regions if the ligand atomic coordinates are not the same. This option serves two purposes:
This approach is fast and can generate perturbation maps in just a couple minutes for ligands over 120 atoms and ligands containing macrocycles or large, saturated, fused ring systems.
It gives full control over the definition of the single and dual regions for each ligand mutation/perturbation.
Perturbation Map (Expert Mode)¶
By default, a perturbation map with redundant edges arranged into loops will be generated. Such a map can be beneficial by both providing better estimates of calculation errors and by reducing certain errors. However, there is a significant computational cost to such a perturbation map (with typical maps increasing the number of mutations carried out by a factor of two or more when compared to the minimum required). Sometimes, for efficiency, speed or cost, a user may wish to instead carry out a star-map calculation, where all mutations are connected to a common ligand by a single mutation (edge). The total number of mutations carried in this case is N-1, where N is the number of ligands. The center of the star map will be the reference ligand, as specified during input.
Max #Charge-Changing Mutations (Expert Mode)¶
Default: 2
This setting defines the maximum number of connections between ligand nodes in the perturbation map that have differing formal charges. This value is described in more detail in the description of the perturbation map below.
2D Ligand Viewer¶
A 2D representation of any ligand that has been imported can be viewed by clicking on the row for that ligand.

Rotate Bond (Expert Mode)¶
In the 2D ligand viewer panel (above), if the user is in Expert mode, it is possible to define a bond for rotation. The rotatable bond, if defined, is a bond for which both the input geometry and a second geometry, rotated 180 degrees about that bond from the input geometry, will be used during the free energy workflow. (If the Rotate Bond values are left blank, this rotation will not be performed). Note that rotations about bonds in the molecule are naturally possible during the molecular dynamics simulation. But for some molecules, there may be a bond for which the rotational barrier is high and so the molecule will not be able to sample sufficiently between conformers separated by that high barrier. In situations where it is ambiguous which conformer is more likely, the Rotate Bond function can be useful.
Start and end are set to the atom numbers (see in 2D plot) defining the rotatable bond. Rotation will be performed with the atoms on the End side of the bond moving, relative to the fixed positions on the Start side. A second conformer is generated by a rotation of 180 degrees about the rotatable bond. This results in two geometries: The starting (input) geometry, and a second geometry reflecting a 180 degree rotation about the selected bond.
The perturbation map (below) will be generated as normal. But when a rotatable bond has been defined for a molecule, all mutations (edges) to/from the node corresponding to that molecule will be carried out twice, once to the starting conformer, and a second time to the 180 degree rotated conformer. Subsequently, the two free energies of mutation for each edge connecting to the node with two conformers will be Boltzmann averaged.
For a molecule where an additional conformer has been defined using the Rotate Bond option, two molecules will subsequently be associated with the molecule (node). The first retains the name as read from the input ligand file (or auto-assigned by the program if no name was given), INPUT_NAME. The second will be named INPUT_NAME-alt. For example, in the case where rotatable bonds are defined for two ligands with the names “ref” and “6e”, the mutations table (described in a section below) would list the mutations list as:

Perturbation Map¶
Once the file uploads are complete, you can create the perturbation map. The perturbation map creates a set of connections (known as perturbations/mutations or edges) among the ligands that define the set of FEP calculations that will be used to evaluate the relative free energies of binding for the set of ligands. The perturbation map must be determined before continuing with the calculation. Once all the input files have been supplied, press the Generate perturbation map button that appears on the bottom right of the File Uploads section. An automated iterative optimization is carried out to define the perturbation map. If the user wants to make changes to the map generated, that is possible using the editing tools (see Perturbation Map Editing).

The perturbation map has two goals:
Evaluate similarity among the ligands, and create connections between ligands that are closely related (for which relative free energy calculations tend to converge more readily)
Ensure that there is at least one connection between every input ligand and other input ligands
With the default map there is a third goal - to use redundant edges and construct closed loops to improve accuracy. This allows the platform to apply our Network Hysteresis Correction (NHC) to the results. As the energy is a state function, the hysteresis calculated from any loops starting and ending at the same ligand should be 0 and any deviation from 0 corresponds to error in the calculations that comprise that loop. The NHC is calculated through linear programming as a series of shifts to the binding affinities to remove the hysteresis across the full perturbation map. The resulting corrections provide estimates of the sampling error and improve the accuracy of the results.
A sample map, for sixteen input ligands, is shown above. The map consists of nodes (shown as circles) and edges (lines that connect the nodes). Each node corresponds to one of the input ligands as defined by the user. The character string next to each node is the name of the ligand (see the Name field of the ligands table). Each edge is a perturbation that will be carried out by the program. The width of the edge reflects the similarity, with thicker edges being more similar, and thin edges being less similar. If you move the cursor over any edge, a number will appear that provides a similarity score, which can range from 0.0 (completely dissimilar) to 1.0 (identical).
You can enlarge/reduce the size of the map using the scroll wheel of your mouse. Enlarging the local map is sometimes useful to better see the varying widths of the edges, or to better read the names next to the nodes.
Very low similarity indices (especially 0.0-0.4) should be examined carefully to ensure that the ligands connected by the edge are acceptably similar to make it possible to determine a reliable free energy. Note that there will usually be more edges (free energy perturbations to calculate) than input ligands. As noted, the additional edges are to reduce the overall error in the net calculated relative free energies.
Hovering the cursor over any node will present the 2D representation of the ligand corresponding to that node.

The user should evaluate the perturbation map carefully before proceeding, ensuring both that the edges make sense, and that the 2D representations are as you expect for the input ligands. Once you are satisfied with both, you can move on to specifying Simulation Options and then running the simulation.
Perturbation Map Progress Bar¶
While the calculations to produce the perturbation map are being carried out, a progress bar will appear at the top of the Perturbation Map section of the panel. When the perturbation map determination is finished, the progress bar will disappear and the map will be shown.

Perturbation Maps with Formal Charge Changes¶
In the case where all the ligands have the same formal charge (e.g. +1, 0, -1, etc.), then the perturbation map is generated as described above. If the ligand set includes ligands with a mixture of formal charges, then in addition to topological and conformational similarity, we must also consider the charge change in optimizing the perturbation map. In this case, the process followed is:
Separate ligands into sets based on their formal net charge
Generate individual perturbation maps for each group of formal net charges
Also generate a perturbation map for all ligands, ignoring formal charge (full set)
Edges from the full set that connect the charge-based sets are introduced (on the basis of similarity and molecular weight) to ensure that there is edge connectivity for all nodes in the map (regardless of charge)
By default, the maximum number of edges connecting nodes with differing charges is 2. This default can be changed in Expert Mode. In Expert Mode, the field where you can change that value appears to the left of the Generate perturbation map button.

Changing the maximum number of charge-changing mutations (perturbations) will have no effect if all ligands have the same formal net charge.
Perturbation Map Editing¶
Using the editing tools, it is possible for the user to clean up the map presentation on the screen and add or remove edges (perturbations) from the map, as described below.
Prettify: This button will automatically adjust the positions of the nodes on the screen to improve the visual presentation. Use this if nodes are overlapped, or if you have adjusted the map edges via editing and want a better view. The Prettify button does not change the underlying simulation matrix. Clicking this button won’t change what perturbations will be carried out–it merely provides a nicer presentation of the matrix on the screen.
Download: This button allows you to download the current view of the perturbation map in the .SVG format.
Edit: This button puts the perturbation map in Edit mode. In Edit mode, you can modify the edges (lines) that connect the nodes (ligands) together. These define the set of perturbations that will be carried out. If you click on the Edit Edge Button, two additional buttons, + and - appear at the lower right region of the region. Click on the Stop button at any time to save the edits you have made and go back into View mode. You can click on Edit again after having clicked on Stop, to make more edits, if desired.
Stop: The Edit button changes to the Stop button when you enter Edit mode. Clicking on the Stop button commits any changes you have made during editing, and returns to the View mode.

To remove an edge, click on the center of that edge. The color of the edge will turn red. Then click on the - button to remove that edge. If you decide you do not want to delete that edge after clicking on it, simply click on another element in the map (another edge or a node), and the previously-selected edge will be de-selected. (You can also click on the Stop Editing button to achieve the same thing).
To add an edge, you need to click on two nodes you want to connect, then click on the + button to add the edge. The order in which you selected the nodes is important: The perturbation direction will be from the first node you select to the second node you select. When you select a node, it will turn red. (If you want to de-select that node, just click it a second time). You cannot add an edge to two nodes that are already connected by an edge. When you click on the first node in Edit mode (Node_1), that becomes the root node for all additions in this edit session. Clicking on a second node (Node_2) then on the + button adds an edge between those two nodes (Node_1-Node_2). Then, clicking on a third node (Node_3) and the + button will add another edge between the first node and the third (Node_1-Node_3). If you wish to pick a different root node for editing, click the Stop button, then click Edit again to start a new set of edits. (The previous edits are saved and retained when you click Stop).
When editing edges, you need to ensure that every node is somehow connected to every other node through a pathway of edges. More generally, to reduce the net statistical errors in the calculated binding energies, every node should be part of at least one loop.
If you create singletons (nodes with no connections) or islands (a group of nodes connected to each other but not connected by any edge to other another island of nodes), the automated workflow will fail. If the ligand set is such that there is no obvious way to connect certain nodes (because islands of nodes are too dissimilar to each other), you may need to set up multiple simulations, one for each island of nodes.
When you are satisfied, click the Stop button. If you make a mistake during editing and wish to revert to the starting map, click on the Generate perturbation map button at the bottom of the file input section. This will delete the current perturbation map and replace it with one auto-generated from the input files.
Star Maps (Expert Mode)¶
If the user has chosen the “Star map” option in the previous section, then instead of the default map with redundant edges to satisfy the NHC map described above, a star map is generated, where one edge connects the reference ligand to every other ligand in the set, as shown below:

This simple map requires the minimum number of perturbation edges to be calculated, and can be useful to save time or compute resources, although at the possible expense of increased overall error. The reference ligand, as specified during input, appears at the center of the star map. All other functions (Prettify/Download/Edit) operate as described for the standard perturbation map above.
Adding Additional Ligands to a Computed Set¶
On occasion, after starting or completing a simulation, you may wish to add more ligands to the set. While you could run an entirely new simulation specifying the larger set, if you have already run calculations for some members of the expanded set, you may wish, instead, to simply add the extra perturbations to those already evaluated to save on computational cost.
You can add additional ligands to a calculation that has either been paused (stopped) or else a calculation that has finished. You cannot add ligands to a job that is in Queued or Running status. If you have submitted a job, and the job has not yet completed, and wish to add additional ligands, click on the Stop button, wait for the status to change to Stopped, add the ligands, and then restart the job.
To add ligands to a stopped or completed job, you simply upload additional ligands via the All ligands (excluding reference) dialog, as described above under File Input Specification. The additional ligands will appear in the list of uploaded structures that appears under the upload dialog, at the end of the list.
After uploading additional ligands, if you subsequently wish to delete any of the added ligands, you can delete them from the list using the red X buttons on each line of the uploaded ligands set. (Be careful not to delete ligands from the original set; if you delete one of those ligands, any calculations related to that ligand will be deleted from the project and cannot be recovered without uploading that ligand again and re-running).
Once you have uploaded the additional ligands, you will see a pop-up that warns you that you will need to regenerate the perturbation map, using the Generate perturbation map button. Clicking on this button will generate a new map that includes the additional ligands, but retains the map that was generated prior to adding the additional ligands. Edges and nodes that include the newly added ligands will be shown in blue. Below, as an example, is the original map generated for a set of ligands, and the updated map that was generated after adding additional ligands. Note that the map for the original ligands may appear slightly different in the new map, but the connections (edges) will be the same. In this example, note that the edge between ligands 17 and 1q is drawn pointing downward in the original map, and it is drawn pointing upward in the map with the additional ligands. This has no effect on the calculation, but aids in presenting a clear view of the map with the added vertices/edges.



By forcing the original map to be retained, the number of additional edges that is generated through this process is minimized and computational cost is kept in check. The resulting map can be edited just as described for the original map, and you can both add and remove edges. Note that if you attempt to remove an edge for a calculation that was already carried out in the first part of the calculation (before adding ligands), the perturbation for that edge will be permanently removed and cannot be retrieved. A warning will appear to avoid inadvertently deleting edges for previous calculations.
Note, also, that because the original map (from before adding additional ligands) is retained in any expanded map that results from adding additional ligands, it is likely that the overall map that is generated when adding ligands to a pre-existing calculation will be different from the map that would be generated if the full set of ligands (from the two steps) had been specified from the beginning. The original map generation attempts to optimize similarity connections while limiting total number of edges. The resulting edges can and will differ depending on the set of ligands provided. Typically, the edge structure of a calculation performed without adding additional ligands later will be overall better optimized. That said, the map generated in two steps will still connect all vertices with at least one edge.
There are a couple of additional limitations when extending the perturbation map:
You can not change the map type (standard vs star map). Whatever option you chose originally will be maintained.
You can not change the parameters in the Simulation Options section (below). These will again be the options you originally chose.
Alternate rotameric states can not be selected for new or existing ligands in the set. This feature also will not work for calculations previously run with alternate rotamers.
After you have uploaded the additional ligands and generated the map (and edited the resulting map, if desired), you can click on the Resume button (below) to restart the simulation for the revised map/ligand set.
The upside of simply adding to an existing calculation is that you reduce the number of new edges that need to be calculated. The downside is that in order to reduce the number of edges added, the resulting map may not be as well optimized as if you had run everything at once.
Simulation Options¶
The options displayed will depend on whether you are running in Standard Mode (default) or in Expert Mode. As noted earlier, Expert Mode, if desired, is turned on in the User Options panel. The standard options will be described first. Expert Mode options will be described in the subsequent section. You will only see one of the FEP Options sets, depending on the status of the Expert selection toggle.
The Overall FEP Workflow¶
The overall FEP workflow, which places many of the options described in the following sections into context, is shown in the following diagram. Here, you can see how the pre-FEP MD equilibrated structure feeds into each lambda window simulation, where it is used as the starting point for additional equilibration and then data collection.

FEP Options (Standard)¶

A limited number of options are provided here. Reasonable defaults are provided for most options related to FEP and are not shown here. The remaining options include:
FEP type: Defines whether this FEP simulation will use only a classical MM force field, or whether this simulation will incorporate a quantum region around the ligand, and use a QM/MM force field.
AI: Use a classical force field for the entire system with ligands parameterized through a machine learning model trained to reproduce QM calculations.
Quantum: Use a QM representation for the ligand and surrounding residues, and a QM/MM force field. The QM representation is GFN1-xTB.
Lambda windows: Also known as lambda points, number of lambda values that connect the two ligands for which a free energy difference is calculated. Convergence is improved by breaking up the free energy simulation into a set of more-similar lambda intermediates and then summing the free energies for those intermediate states to get the total free energy. One trajectory is run per lambda window. The default is Lambda windows = 16 for AI, and Lambda windows = 14 for quantum. The windows are denser near the endpoints to improve convergence.
Simulation time (ns): The amount of molecular dynamics (MD) sampling done for each FEP window to obtain a converged value of the quantity that determines the free energy change for the corresponding lambda window. The default value is 5.0 ns for an AI simulation, and 2.0 ns for a Quantum (QM/MM) simulation.
Reset to default: Reset all options to their defaults.
Start simulation: This option will start the FEP simulation. This option only appears for FEP Type = AI. For FEP Type: Quantum, additional options related to the quantum region need to be set after system preparation. If you need to set the calculation to pause after the Preparation stage for an AI simulation, tick the box next to Stop after preparation (found above this button).
Start preparation: This option will start FEP preparation. This option only appears for FEP Type = Quantum, or if Stop after preparation was ticked (see below). For FEP type: AI, stopping after preparation is not typically necessary and the preparation and simulation steps are combined.
Stop after preparation: Pauses the workflow after the preparation step. This is the default (and can’t be changed) for FEP type: Quantum, because the user must subsequently define the quantum region. For FEP type: AI, the user can force a pause in the workflow by clicking this box.
FEP Options (Expert)¶
If you have enabled Expert Mode in the User Settings panel, then a larger number of FEP options are accessible. These options will appear below the perturbation map.

FEP type: Defines whether this FEP simulation will use only a classical MM force field, or whether this simulation will incorporate a quantum region around the ligand, and use a QM/MM force field.
Classical: Use a classical force field for the entire system with ligands parameterized traditionally based on a short QM calculation
AI: Use a classical force field for the entire system with ligands parameterized through a machine learning model trained to reproduce QM calculations.
Quantum: Use a QM representation for the ligand and surrounding residues, and a QM/MM force field. The QM representation is GFN1-xTB.
Quantum (GFN2-xTB): Use a QM representation for the ligand and surrounding residues, and a QM/MM force field. The QM representation is GFN2-xTB, which differs from the GFN1-xTB with the inclusion of anisotropic second order density fluctuation effects. This level of theory can especially help with treatment of metal interactions.
Random number seed: A random number that will be used to create a chain of random numbers. These random numbers are used to set the initial velocities for the system. Using a different random number will create a different set of velocities and a different overall trajectory. A value of -1 uses the default random number seed, which is based on the current Unix time when the job is submitted. Submitting two jobs with the default value of -1 and no other differences would generate two trajectories that vary due to their stochastically-generated initial velocities. If you wish to be able to exactly reproduce the same results when running the same job twice, you need to specify a (non-default) random number seed. E.g. running the calculation twice with a random number seed of 32177 would give identical results.
pre-FEP equilibration (ns): The total amount of MD equilibration to perform before starting the FEP runs, in nanoseconds. This value does not include any MD requested by Heating Duration and Pressurization Duration (below). The default is 0.25ns (250ps). This value also does not include any additional QM/MM equilibration (below) that is performed in the case of a Quantum simulation.
Heating duration (ns): The temperature will be ramped up from the initial temperature of 0K to a target temperature of 300K over the specified number of nanoseconds of simulation. This Heating Duration ramping time is not included in the total Pre-FEP Equilibration time (above). The default is 0.1ns (100ps).
Pressurization duration (ns): The pressure will be ramped up from the value calculated from the input system to the Target Pressure of 1atm over the specified number of nanoseconds of simulation. This Pressurization Duration ramping time is not included in the total Pre-FEP Equilibration time (above). The default is 0.1ns (100ps).
QM/MM equilibration (ns): For a QM/MM simulation, additional pre-FEP equilibration is generally applied to more thoroughly allow the system to reflect the force field. The QM/MM equilibration is performed using the QM/MM force field that will also be used for the FEP calculation. QM/MM equilibration is not applied for Classical or AI mode. For QM/MM, total equilibration is therefore: (Pre-FEP Equilibration) + (QM/MM equilibration). The default is 1.0ns.
The heating ramp is performed before the pressurization step, after which the Pre-FEP Equilibration MD is performed. Total FEP Pre-Equilibration simulation time will be:
(Default is 450ps total for a classical or AI simulation, 1,450ps for a QM/MM simulation).
The remaining options define the number of intermediate lambda windows, and the amounts of equilibration and data collection performed at each window. The defaults are set to widely-accepted values used in scientific literature. A full free energy calculation consists of two separate calculations, one for the ligand bound to the receptor, and a second for the ligand free in solution. The values defined here will be used for both separate calculations.
The FEP simulation at every lambda window starts with the equilibrated system as generated according to the Pre-Equilibration Options above. Additional equilibration is performed using a potential energy function that reflects that specific lambda window. The amount of additional equilibration to perform is defined by the Equilibration (ns) parameter below.
Lambda windows: Also known as lambda points, number of lambda values that connect the two ligands for which a free energy difference is calculated. Convergence is improved by breaking up the free energy simulation into a set of more-similar lambda intermediates and then summing the free energies for those intermediate states to get the total free energy. One trajectory is run per lambda window. The default is Lambda windows = 13 for Classical, Lambda windows = 14 for Quantum, and Lambda windows = 16 for AI. The windows are denser near the endpoints to improve convergence.
Random number seed: A random number that will be used to create a chain of random numbers. These random numbers are used to set the initial velocities for the MD used in generating the FEP statistics. Using a different random number will create a different set of velocities and a different overall trajectory. A value of -1 uses the default random number seed.
Equilibration (ns): The number of nanoseconds of MD equilibration to perform at each lambda window before data collection starts. Default is 0.5 ns.
Simulation time (ns): The amount of molecular dynamics (MD) sampling to perform for each lambda window to obtain a converged potential energy trajectory used to determine the corresponding free energy change for that window. The default value is 5 ns for a Classical or AI simulation, and 2 ns for a Quantum (QM/MM) simulation.
Check for buried waters: Performs a Grand Canonical Monte Carlo (GCMC) FEP simulation, where insertion and removal of buried water molecules will be periodically attempted during the simulation.
Use mixed precision: Use mixed precision rather than double precision floating-point number format during the calculation. This is an option when running on GPU hardware and greatly increases the throughput of the calculation.
Reset to default: Reset all options to their defaults.
Stop after preparation: Pauses the workflow after the preparation step. This is the default (and can’t be changed) for FEP Type = Quantum, because the user must subsequently define the quantum region. For FEP type = Classical or FEP type = AI, the user can force a pause in the workflow by clicking this box.
Runtime Options¶

This section gives control over the computational resources used during the simulation.
Cloud option: Selects the cloud computing priority for this calculation. On-Demand is the highest priority, and once the calculation is started, it will not be pre-empted. It guarantees the fastest (wall clock) throughput. Spot-Instance is a lower priority, and can be pre-empted by the cloud system if there is high demand from competing jobs on the cloud platform. Calculations run with Spot-Instances can have a significantly lower cost, but jobs run using Spot-Instance may take around 2-3x longer to complete. It is possible to change the computing priority for a calculation that has already been submitted. To do this, Stop the job in the Control Panel (see options under Simulation Status), change the priority, then Restart the job. The change will apply to the portions of the calculation that have not already completed. This option only applies to jobs run on shared cloud resources; it does not apply to calculations run on a local HPC cluster, or on reserved resources in a cloud environment.
FEP Preparation¶
Ligand Alignment¶
The first stage in preparation is to accurately align the ligands to allow the automated determination of topological correspondences required for FEP calculations. The progress of this preparation portion of the workflow is indicated by a progress bar, as shown.

QM Region Specification (Mutations Where Formal Charge Is Not Changing)¶
If you have set FEP Type = Quantum in the FEP options, then an additional panel will appear below the FEP Options section. If you are running a FEP Type = Classical or AI simulation, the entire system is treated using MM, and this section is hidden.
The QM region specification options are described in more detail in the section qm-region-selection. Here, a simple description of the options is provided.
The platform separates out mutations (perturbations) into two classes: Those where the net (formal) charge of the ligand remains constant during the perturbation, and those where the net (formal) charge of the ligand changes during the mutation. These classes are treated separately, in terms of defining the QM region. This is because it is often found that when the net charge of the ligand changes, a somewhat larger definition of the QM region is useful to improve accuracy. The platform automatically selects QM regions (separately) for the two classes, but using the options here you can modify those default selections.

In this section, the user can specify the extent of the region to be treated at the quantum mechanics level. The MD calculations are carried out using a QM/MM implementation, where the core residues (the ligand and chosen surrounding residues) are treated with the higher accuracy QM approach, and the remainder of the system is treated using a classical molecular mechanics force field.
For QM/MM simulations, the ligand must always be included in the QM region. The ligand is represented by the keyword “ligand” (lower case, no quotes) in the definition box.
Residues included in the QM region are filtered by the maximum number of atoms in that region. The maximum atoms filter is applied when generating an automated definition of the QM region (Suggest a QM region), along with a cutoff distance of 5 Angstroms. The distance between the ligand and a protein residue is defined as the shortest distance of any atom of the ligand to any atom of that residue. If that distance is less than 5 Angstroms, the residue is a candidate for being included in the suggested QM region.

Region Max Atoms: The maximum number of atoms that will be included in the QM region if the region is automatically generated using the Suggest a QM Region button (below). The atom cutoff is implemented on a residue basis, so that any residue is either entirely within the QM region, or it is excluded from that region. The default is 100 atoms. Increasing the number of atoms in the QM region will increase the computation required for the simulation to finish.
Selection Algorithm: [Simple/Optimal] This selection applies when using the automated “Suggest a QM Region button (below). In both cases, an initial set of protein residue candidates for inclusion in the QM region is generated based on a cutoff distance of 5A from the ligand. A ranking value is assigned to every residue candidate, and then residues are removed from the candidate pool, based on rank, until the Region Maximum Atoms limit is met.
Simple: The ranking value for each protein residue in the candidate set is based on the minimum distance of any atom of the protein residue to any atom of the ligand. A higher ranking value corresponds to a larger distance. Residues are removed from the candidate pool in order of ranking (high to low).
Optimal: The ranking value for each protein residue in the candidate set is based on both the minimum distance of any atom of the protein residue to any atom of the ligand and whether the protein residue makes specific close-distance polar interactions with the ligand. A higher ranking value corresponds to a residue making more significant interactions. Residues are removed from the candidate pool in order of ranking (high to low).
The text input area: In the middle of this region, you will find a text input box. You can specify residues that are to be included in the QM region in this text box using the format described in the chapter “QM Region Specification.” This box can be populated either manually, by writing in the definitions on your own, or else automatically by pressing the Suggest a QM Region button. The ligand must always be included in the QM region, using the definition “ligand” (lowercase, no quotes). If you use the Select a QM Region button, the ligand will appear at the top of the suggestion text automatically. Note that there are two tab buttons above the text input area. The Charge Conserving tab shows the QM region definition for perturbations where the net formal charge of the ligand is not changing. If there are any perturbations where the net formal charges of the ligand does change, then the Charge Changing tab is active, and you can push on that to similarly define the QM region for that that type of perturbation (shown in the next section.) If no perturbations have a net change in formal charge, the Charge Changing tab is not selectable.
Suggest a QM region: Auto-populates the text input area with a set of residues that meet the criteria described Selection Algorithm (above). The user can modify the list once it is generated (adding or subtracting residues).
Validate: This button allows the user to validate the set of residues that are specified in the text input area above the button. Validate will ensure the residue definitions correspond to residues in the input PDB file. Validate will also add capping residues, if necessary, to ensure proper QM/MM boundaries for the calculation. If you use the Suggest a QM Region button and do not modify the contents of the definition box, the contents are automatically validated, and the Validate button is inactive. Only if you change the contents of the definition box will the Validate button be active.
Visualize: Creates a pop-up window that shows the selected QM-region residues in the context of the protein/ligand complex. The reference ligand will be shown in blue, bound to the protein. The protein residues included in the QM region will be shown in red. You can rotate the view using your mouse (left button) or translate the view with the mouse (right button). Double-click on any atom to reset that atom as the center for rotations. If you hover the mouse over any atom, information on the atom and the associated residue (name, number) will pop up. To close the pop-up window, click the X near the top right of the box.

Charge Conserving QM Region Summary: To the right of this section, you will find the QM Region Summary, which is populated after you validate the QM region contents. (The definition is auto-validated, and this region is auto-populated if you use the Suggest a QM Region button). In this section, you will find:
Status: Whether the contents of the definition box properly validated. The possibilities are Validated or Invalid. If the contents are Invalid (there is a problem) or if they are valid but a warning was issued, the status will be shown in red, and you can ascertain more information by hovering the cursor over the question mark.
Number of link atoms: Number of hydrogen atoms that were added to address unfulfilled valencies that arose from using separated protein residues. These are required for the QM simulation.
Number of total QM atoms: Total atoms in the QM region, excluding the ligand itself. Based on the reference ligand.
Total QM charges: Total QM-determined charge in the QM region (ligand + other residues), based on the reference ligand.
QM Region Specification (Perturbations Where Formal Charge Is Changing)¶
As noted earlier, the platform separates perturbations into two classes, those where the formal charge of the ligand does not change, and those where the formal charge of the ligand does change. The controls in the QM Region Specification section are similar for the two cases, but provision is made to allow the user to modify and visualize the QM regions for the two classes separately. To view the definition for charge-changing perturbations, click on the Charge Changing tab above the center text box, as show below:

When you click on this tab, you’ll note that the header on the information to the right changes from “Charge Conserving QM Region Summary” to “Charge Changing QM Region Summary”
This tab is not selectable in cases where there are no perturbations for the system where the net formal charge changes.
The following two options are applicable only to charge-changing perturbations. All other functionality works identically as with non-charge-changing perturbations, as described above.
Region Max Atoms (Charge)
The atom maximum here applies only if there are perturbations between residues that have differing net formal charges. If there are no perturbations between nodes with differing formal charge, then this option will be greyed out. The QM region for perturbations where a formal charge change occurs is selected via a different approach than the remainder of the perturbations. For formal charge change perturbations, the chosen QM region is informed by QM-based polarizability calculations on the binding site. Typically, a somewhat larger maximum number of atoms is required for these perturbations. With this option, you can set the maximum number of atoms for charge change perturbations (or accept the default).
Algorithm (Charge)
The algorithm selector here applies only if there are perturbations between residues that have differing net formal charges.
Currently, this option is limited to one choice, which is pre-selected and cannot be changed:
Optimal: The ranking value for each receptor residue in the candidate set is based on both the minimum distance of any atom of the receptor residue to any atom of the ligand and a set of semiempirical QM calculations that assess how sensitive the charge distribution of each nearby receptor residue is to the changes in charge during the ligand perturbations. For all receptor residues that pass the distance check, a set of calculations is performed to assess the average magnitude change of charges in the that residue with each perturbation in the perturbation map. The surrounding residues are then ordered, from largest overall change to smallest, and the residues with the largest changes are included in the QM region until the maximum total number of residues (above) is reached.
Quantum Region Selection when RNA or DNA Is Present¶
The QM region selection fully supports nucleotides in addition to proteins. You can still generate suggested QM regions. The selection types are different, however. See the chapter on qm-region-selection for more details.
Start Simulation¶
Once you are satisfied with your quantum region selections, you can start the simulation using the Start Simulation button at the bottom right of this part of the panel.
Simulation Status¶
This portion of the panel provides the status of the calculation once it has been started using the Start Simulation button. It also provides the ability to stop and restart a calculation that has been submitted.

Stop: Stop a calculation that was previously submitted and is in progress. A stopped calculation is saved in the cloud storage associated with your account, and can be restarted later, using the Run command.
Run: Run a previously stopped job. If the job has not previously been started using Start Simulation, then the Run button has the same effect as Start Simulation, i.e., it will begin the calculation.
Cloud option: Changes the cloud computing priority for this calculation. On-Demand is the highest priority, and once the calculation is started, it will not be pre-empted. It guarantees the fastest (wall clock) throughput. Spot-Instance is a lower priority, and can be pre-empted by the cloud system if there is high demand from competing jobs on the cloud platform. Calculations run with Spot-Instances can have a significantly lower cost, but jobs run using Spot-Instance may take around 2-3x longer to complete. It is possible to change the computing priority for a calculation that has already been submitted. To do this, Stop the job in the Control Panel, change the priority, then Restart the job. The change will apply to the portions of the calculation that have not already completed. This option only applies to jobs run on shared cloud resources; it does not apply to calculations run on a local HPC cluster, or on reserved resources in a cloud environment.
Below, and also to the right of the control buttons, you will find information about the status of your job. The total estimated virtual CPU usage (vCPU) is given, as are progress bars for each of the stages of the simulation: Preparation/Equilibration/Production/Analysis. Color coding is as follows:
Light Grey: That segment of the calculation workflow has not been run.
Dark Blue + Light Grey: That segment of the workflow is in progress, and the dark blue portion of the segment reflects a progress bar.
Dark Grey: That segment of the workflow has completed.
Red: That segment of the workflow completed with errors.
Further down in this portion of the page, the status is provided at a more detailed level, showing the progress for every ligand perturbation (every perturbation represented by an edge in the perturbation map. The names of the starting and ending ligands for each perturbation are shown in the table, along with a four-segmented progress bar. The four segments for the progress bar represent Preparation/Equilibration/Production/Analysis. Each segment is colored as described above.
Diagnostic Report¶
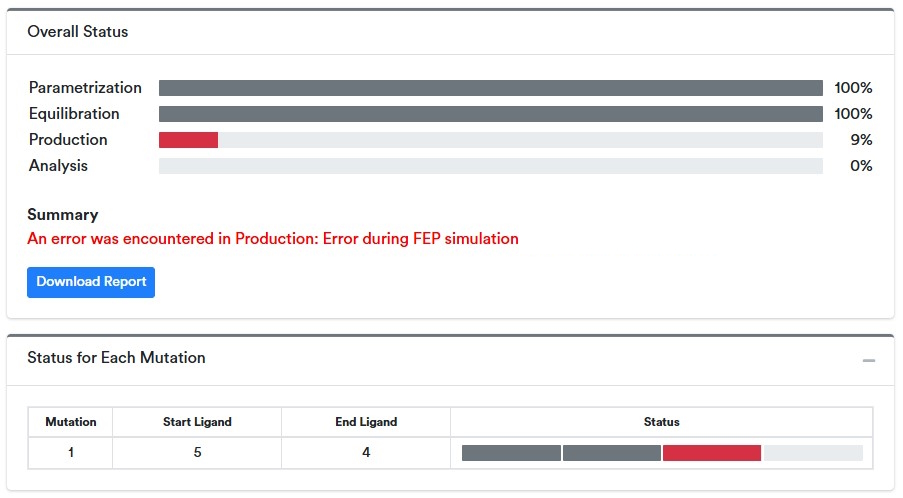
Any issues encountered during runtime are reported in the Simulation Status section. These can arise from how the inputs were prepared or from the choice of settings, particularly if they deviate significantly from the defaults. Often a simulation that fails due to a transient event (hardware or software) can be successfully resolved by resuming the calculation using the “Resume” button. If a calculation continues to fail immediately after resuming, then it is best to check the summary and diagnostic report for details to aid resolution.
When an issue occurs, a red message will appear under the Summary header, as well as a Download Report button. The affected mutation will appear red.
Pressing the Download Report button will generate and download two diagnostic reports. The first contains a log file with traceback information and further details of how and when the issue occurred. The other, labeled “with_structures” also includes the initial and most recent structures at the point of termination. This information is sufficient to identify common failure modes and to aid in making modifications to the calculations that circumvent them. In other cases, forwarding this information to your IT or to QSimulate’s support team can aid in efficiently identifying the issue.
Results¶
Below the Simulation Status section, you will find the results of your calculation. The information in this section will be auto-populated when the simulations requested have completed. The data is provided in the following sections:

Result for Each Mutation¶
One result will appear in this table for every edge in the perturbation map. The edges (lines) represent pairs of ligands between which free energy calculations will be carried out. The number of connections (edges) will typically be larger than the number of input ligands. The relative free energy changes for all the edges will be calculated. Performing more than the bare minimum number of changes required to connect the ligands helps provide more reliable net free energy difference values and associated errors in the Free Energy Results table (below).
All columns of the Result for Each Mutation table are sortable. Clicking on the header once will sort low-to-high. Clicking on the header a second time will sort high-to-low.
For each mutation (perturbation) that was run, the following will be reported:
Mutation: Sequence number for the mutation calculation, starting from one.
Start Ligand: The name of the starting ligand for the mutation. The name is either taken from the user input ligand file or, if not present in that file, assigned by the program as LNNN, where “NNN” is the counter for ligands without user-supplied names, which starts at 1. These names are auto-assigned, if necessary, for ligands supplied in the All ligands (except reference) input. For example, the first ligand read in without a name would be L001, and the tenth ligand read in without a name would be L010.
End Ligand: The name of the ending ligand for the mutation. The name is obtained in the same fashion as for Start Ligand.
ddG (BAR): The computed free energy of binding (in kcal/mol) for the specified mutation between the start and end ligands. ddG is calculated as the difference [dG(complex) - dG(ligand)] using the Bennett Acceptance Ratio approach, which provides the net free energy of binding via the thermodynamic cycle relationship.
ddG (Solv): The ddG of solvation by calculating the dG in vacuum from the ensemble of unbound ligand structures and comparing to the dG ligand. This indicates whether a mutation makes a ligand more hydrophobic or hydrophilic. A negative value means that the end ligand (lambda = 1) of that mutation has a better solvation energy - it is more stable in solvent. If a ligand that is a strong binder has a lower or comparable solvation energy to other ligands in your set, its binding to the target is more likely to be driven by specific interactions and therefore suggests greater specificity for the target. Conversely, a strong binder that has a more unfavorable (higher) solvation energy than other ligands in your set may be driven by just increased hydrophobicity and have lower specificity.
Quality Index: A quality index that captures the overlap in the energy ensembles between lambda windows. An index of 0.5 or greater indicates a sufficient overlap and good sampling. A poor index (<0.0) suggests that more lambda windows may be need for this calculation.
Clicking on the triple dot menu to the right of each mutation reveals pop up pages that can be selected for additional information. Each window is covered in the later sections.
Below the Result for Each Mutation and the Free Energy Results tables, there are Export table buttons.
Export table: Download the data in the table as a CSV file.
Stability Analysis¶
This page contains information to assess whether the perturbation calculation is converged and stable.

2D representations of the ligands corresponding to the two endpoints of the mutation are presented at the top of the pop-up window. There are also the energies for the mutation reprinted here, alongside two additional energetic properties:
dG complex: The free energy (kcal/mol) for the mutation between the start and end ligands calculated in the ligand/protein complex.
dG ligand: The free energy (kcal/mol) for the mutation between the start and end ligands calculated in free solution.
Below the table of energies are a series of four plots:
Accumulated free energy: This graph presents the accumulated ddG free energy differences versus lambda for the transformation selected in the Mutations table. The lambda values for the physical endpoints are always 0 and 1. Values are presented for the net ddG = [dG(complex) - dG(ligand)] at each value of lambda. Rolling the cursor over any point of the plot will show the (x,y) values for that point.
Convergence with respect to sampling: This graph presents how the calculated net free energy for the mutation would vary if we used only a subset of the collected data, up to the maximum amount of data collected. From this curve, one can infer how well converged the results may be, and whether the sampling was sufficient. Rolling the cursor over any point of the plot will show the (x,y) values for that point.
RMSD plots: These two plots, one for each ligand, capture the evolution over the simulation of the RMSD of the ligand in the complex bound state. The reference is the input structure. The plots are drawn from the simulations of the first and final lambda windows, representing each ligand when it is fully present, rather than one of the intermediate, alchemical states.
Interaction Analysis¶
This page contains information about the most critical interactions at a glance.
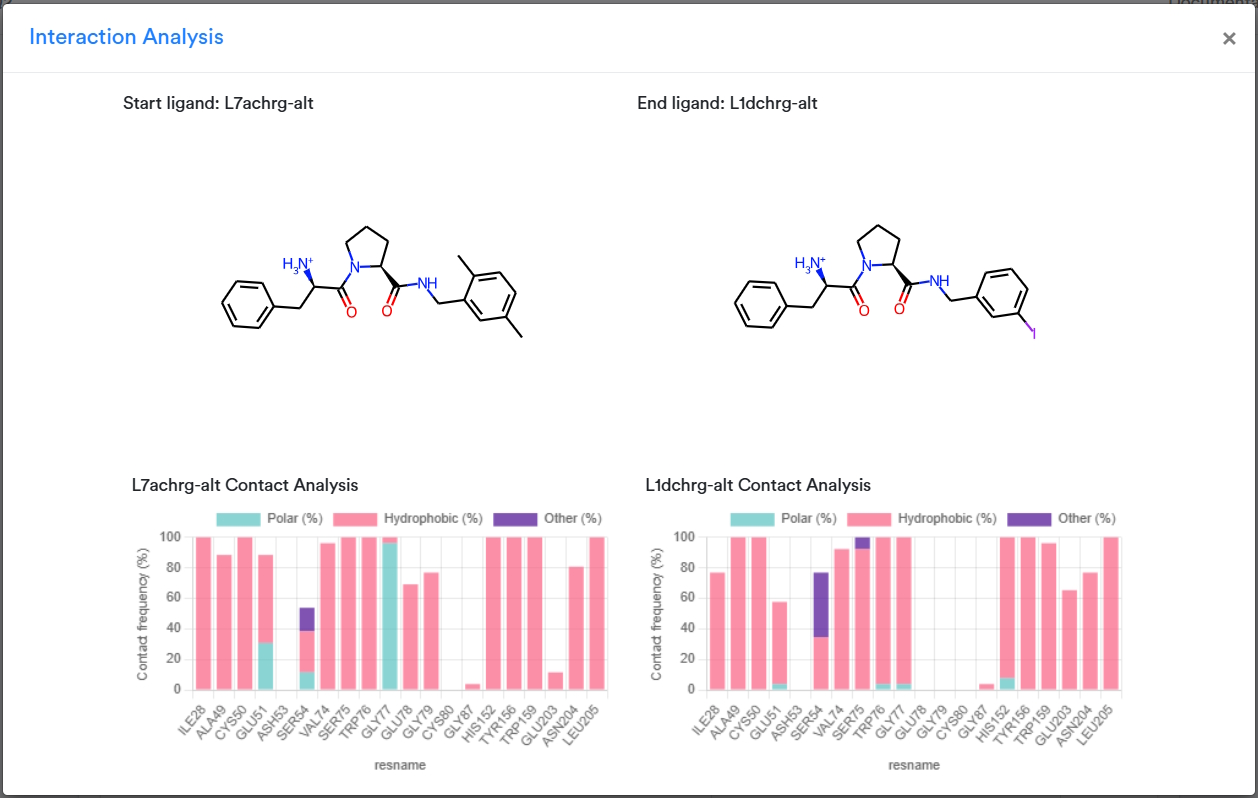
2D representations of the ligands corresponding to the two endpoints of the mutation are presented at the op of the pop-up window. There are two plots with the overall Contact Analysis, one for each ligand. These plots are drawn from the simulations of the first and final lambda windows, representing each ligand when it is fully present, rather than one of the intermediate, alchemical states. Each plot captures the frequency of contacts between the ligand and the target during the course of the full simulation, classified into categories of polar, hydrophobic, and other interactions.
Download Analysis Report¶
This automatically downloads a directory containing detailed reports with trajectory analysis for each lambda window for the selected mutation. Reports for both the complex and ligand legs of the calculation are included. The reports include plots over time of the target RMSD, ligand RMSD, RMSF, dihedrals, and contacts as well as analysis of metal interactions if a metal is present near the ligand. Representative 2D interactions plots showing each ligand’s significant interactions with the target can be found in the report. Also included is the raw data, in csv format, used to generate each plot in each report.
Download Trajectory¶

Preparing the trajectory for download takes a short while, and the user has the option to cancel the download during this process. Once prepared, the download can either be performed in the tab itself or by copy-pasting the S3 link into another tab.
The trajectories are available as DCD files with a PDB topology. There is a DCD file for each window in the bound complex and unbound ligand states. These files can be viewed with external tools like VMD and Pymol - the topology file must be opened first, followed by the DCD file. Note that the default behavior in most visualizers is to insert the topology file structure as the first frame, so this should be ignored in any analysis of the trajectory. Note as well that both ligands of the mutation are present in every trajectory, so some visualizers may initially draw bonds inbetween the two ligands. This can be resolved in the external viewer by hiding one ligand and viewing just the other.
Free Energy Results¶
In this table, the net relative free energies for all the input ligands are presented. The net free energies are evaluated by averaging the energies for multiple paths (edges) that connect the ligand to the reference ligand. The Sampling Errors come from the hysteresis, where applicable, through the NHC.
All columns of the Free Energy Results table are sortable. Clicking on the header once will sort low-to-high. Clicking on the header a second time will sort high-to-low.
Ligand: The name of the ligand. The name is either taken from the user input ligand file or, if not present in that file, assigned by the program as LNNN.
ddG: The free energy of binding for the ligand (kcal/mol), relative to the reference ligand. The reference ligand is arbitrarily assigned a free energy of binding of 0.00. Relative free energy values ddG should be interpreted as ddG = [dG(ligand) - dG(reference)]. A negative value means a stronger binder than the reference ligand.
Sampling Error: Sampling error (kcal/mol) is estimated with NHC. If a mutation is not part of any loop defined by edges, then the estimated error is obtained from the error of the nearest ligand on a loop. If there are no loops (e.g., a task using a star map), the error is obtained from a combination of the statistical analysis carried out during the BAR processing for that mutation (energies calculated in ascending and descending direction for each lambda window). The sampling error is to be considered a lower bound.
Clicking on any row of the Free Energy Results table will show the 2D structure of the ligand for that row, on the right-hand side of the panel.
Result Download¶

At the bottom of the panel, you will find the Result Download button. You can use this button to download a ZIP format archive, named BATCH_NAME_raw_outputs.zip. (BATCH_NAME is the name you have given this batch calculation; For example, if your Batch was named FEP1, then the file would be named FEP1_raw_outputs.zip). Within this archive, you will find the following directories:
BATCH_NAME_raw_outputs.zip > datafile > input > ligands
BATCH_NAME_raw_outputs.zip > datafile > input > receptor
BATCH_NAME_raw_outputs.zip > datafile > ligand_output
BATCH_NAME_raw_outputs.zip > datafile > mutations
ligands subdirectory: Contains a set of subdirectories with the input ligand structures, as provided by the user.
receptor subdirectory: Contains the input protein receptor structure, as provided by the user.
ligand_output subdirectory: Contains a CSV format file with the calculated free energies, one line per input ligand.
mutations subdirectory: Contains several entities.
mutation_results.csv is the calculated relative free energies for each of the mutations (edges) defined by the mutations map, in CSV format.
specific mutations subdirectories: Each of these mutation subdirectories has a name of the format ligand1__ligand2, where ligand1 and ligand2 are the user-supplied (or program auto-generated) names. There is one such directory for every edge in the mutations map. Data within ligand1__ligand2 subdirectory is for the FEP calculation between nodes ligand1 and ligand2.
Within each of these mutation (edge) ligand1__ligand2 subdirectories you will findbound (directory)unbound (directory)convergence_trajectory.csvlambda_trajetory.csvThe two CSV files contain the data used to produce the convergence and accumulated free energy vs. lambda plots that are presented when you click on any of the mutations in the GUI.
Within both the bound and unbound subdirectories will find a series of additional subdirectories, one for each lambda window used for that FEP calculation, each namedlambda_NNNwhere N ranges from 001 to the number windows used. Within each of these subdirectories, you will find a PDB format file
final.pdbthat corresponds to the entire system (ligand/protein/waters/cofactors/ions) on the final step of the MD sampling trajectory used for that window. This file can be read into any standard molecular visualization program.
In both the bound and unbound subdirectories, at the top level of that subdirectory, you will also find the fileinitial.pdb, which is the starting structure of the entire system (ligand/protein/waters/cofactors/ions) at the start of that window calculation.
Covalent FEP¶
QUELO is capable of performing covalent FEP. Covalent FEP can be used to calculate the binding affinities of reversible covalent inhibitors or irreversible covalent inhibitors for mutations away from the warhead. It follows a similar thermodynamic cycle to regular FEP, but with the bound state involving the ligands covalently bound rather than non-covalently bound to the target.
In the case of a covalent FEP task, the only main difference is in the file upload section. The rest of the calculation will proceed regularly, with two exceptions:
currently, only quantum FEP is available for covalent FEP
the quantum region is automatically selected to be the ligand and the side chain of the residue to which it covalently attaches, and this choice cannot be changed.
The covalent FEP File Uploads section is pictured below:

Reaction Type¶
The type of the reaction the ligand has with the protein. Currently, four reaction types are available:
Michael Addition, residue: cysteine, warhead: alpha-beta unsaturated carbonyl with C=C double bond
Thiol-aldehyde Addition, residue: cysteine, warhead: aldehyde
SN2, Chloroacetamide, residue: cysteine, warhead: chloroacetamide
Vinyl-sulfone Cysteine Inhibition, residue: cysteine, warhead: vinyl-sulfone
Imine Formation with Lysine, residue: lysine, warhead: aldehyde
Residue Code¶
The code of the residue that is covalently bound to the ligand. For example, in the screenshot above,
A:CYS:12
refers to chain A, residue number 12, a cysteine.
Protein receptor¶
The PDB file of the protein receptor. This file should not include the covalently bound ligand. The protonation state and any other chemical changes to the reacting residue will be automatically resolved by the platform.
Reference ligand¶
The reference ligand’s coordinates close to its the covalently bound state. The product structure will be automatically generated by the platform so long as the warhead is close to the reacting residue specified by the Residue Code section.
All ligands (excluding reference)¶
A list of ligands, either in SDF or MOL2 format. These coordinates will be aligned to the reference ligand (unless the user selects “perfectly prealigned”), as in the case of non-covalent FEP.
Preparing a PDB File for Use with QUELO¶
Introduction¶
The most common file format used for large molecule coordinates is “PDB.” The legacy PDB format has undergone several revisions over the years, but the fundamental elements have not changed. The legacy PDB format is a fixed format, with a set of descriptors for each atom line in specific columns. A detailed description of the format is available from the Protein Data Bank: Legacy PDB format .
Protein structure files downloaded from the PDB repository will generally be in this format (with a suffix of .PDB or .ent). Generally, macromolecular structures determined by all common structure determination approaches (X-ray crystallography, Cryo-EM, NMR) are deposited in this format.
Preparing a PDB File for QUELO¶
While the general format of these files is standardized, if you wish to use a PDB (or ENT) file for computation, it must generally be cleaned up before the first use. Clean-up of a PDB file that may need to be performed may include (and may not be limited to) addressing the following:
Removal of multiple replicates of the protein in the unit cell due to space-group symmetry
Removal of unnecessary cofactors
Removal of bulk waters
Removal of unsupported metals/cations/anions (see “Supported atom types” below)
Adding missing hydrogens
Determining the protonation state of residues where multiple protonation states are possible at physiological pH (e.g., His)
Determining the proper rotamers of rotatable sidechains that can make hydrogen bonds
Addition of non-hydrogen atoms that may be missing in the structure (due to insufficient resolution or conformational variability)
Ensuring residue names are compatible with the computational platform
Addressing residues with non-standard names
Ensuring that disulfide bonds are properly reflected (using CYX for the parent residue names)
Ensuring that the PDB format is adhered to (the required information for each line is provided in the correct columns).
How a user chooses to address these various questions will depend on the particular system. While some general-purpose routines can be developed to tackle some of these questions, in general, the user is urged to spend some time ensuring that they are starting with a model of the protein receptor (and cofactors, if any) that is sufficient for their needs. A little time spent properly assessing and cleaning up the starting structure can pay significant dividends in terms of better computational results.
QUELO expects that the PDB input file is structurally complete and adheres to the Amber forcefield format. The format that QUELO expects for PDB input files is described below, alongside general information for PDB files.
QSimulate also offers an automated tool called ProtClean to prepare PDB files. For more information, see its chapter in the manual - ProtClean: The Protein Preparation Tool. Any PDB file that will be used as the target receptor in QUELO must be processed through ProtClean. ProtClean will verify and reformat any PDB file to be suitable for QUELO, and will, optionally, perform preparation functions.
Understanding the PDB File (Basics)¶
Every line in a legacy PDB file starts with a line-type designator in the first six columns. Protein atoms typically start with ATOM in this field. Non-protein atoms often use HETATM in this field, although some files will use ATOM for these atoms as well.
A sample PDB format line is shown below:
ATOM 90 NH1 ARG A 11 35.920 5.701 50.185 1.00 54.09 N
123456789 123456789 123456789 123456789 123456789 123456789 123456789 123456789
A description of the contents of the various fields in a typical PDB file line is given below. Note that some of these elements are not used in many files, and some of these fields, in particular the fields that follow the x/y/z coordinate fields, are sometimes used to hold other information–although this is not so common for files that are deposited with the PDB.
PDB file fields
--------------------------------------------------------------------------------------
Field | Columns | Format | Required |
| | | by QUELO | Contents
1 | 1 - 6 | A6 | YES | Record Type (Atoms use ATOM or HETATM)
2 | 7 - 11 | I5 | YES | Atom Sequence Number
3 | 13 - 16 | A4 | YES | Atom Name
4 | 17 - 17 | A1 | NO | Alternative conformation flag (optional)
5 | 18 - 20 | A3 | YES | Residue Name
6 | 22 - 22 | A1 | NO | Chain Identifier (optional)
7 | 23 - 26 | I4 | YES | Residue Sequence Number
8 | 27 - 27 | A1 | NO | Insertion Code (optional)
9 | 31 - 38 | F8.3 | YES | X-coordinate
10 | 39 - 46 | F8.3 | YES | Y-coordinate
11 | 47 - 54 | F8.3 | YES | Z-coordinate
12 | 55 - 60 | F6.2 | NO | Occupancy value for atom (optional)
13 | 61 - 66 | F6.2 | NO | B-value (thermal factor; optional)
14 | 67 - 76 | | NO | Various use
15 | 77 - 80 | A4 | YES | Atom type. Right justified.
--------------------------------------------------------------------------------------
The fields that QUELO expects to find for each record are shown in the table above. The alternate conformer signifier, if found, specifies that there are two or more conformations that contribute to the atom at that position. In this case, QUELO will automatically take the first position. The fields indicated as not Required by QUELO can still be specified but won’t necessarily be used.
Protein Residue Naming Convention Used by QUELO¶
The formal protonation/charge state for each protein residue is inferred from the residue name. It is important that you prepare your protein with this in mind. Protein structures obtained from sources like the PDB do not always adhere to these conventions. For most standard amino acid residues, there is only one variant that exists at physiological pH. But for a few residues–ASP, GLU, LYS, and HIS–multiple charge states can exist within the range of physiological pH, and depending on the local environment. The residue name convention used by QUELO (and atom naming conventions associated with each residue type) follow the conventions below:
Residue Name | Charge State | Notes
ARG | +1 |
ASP | -1 |
ASH | 0 | Neutral (protonated) ASP
CYS | 0 |
CYX | 0 | Neutral as a part of a disulfide bond
CYM | -1 | Charged (deprotonated) CYS
GLU | -1 |
GLH | 0 | Neutral (protonated) GLU
HIS | 0 | Defaults to HIE (below)
HID | 0 | Hydrogen on delta N
HIE | 0 | Hydrogen on epsilon N
HIP | +1 | Hydrogens on both delta and epsilon N
LYS | +1 |
LYN | 0 | Neutral (deprotonated) LYS
ALA | 0 |
ASN | 0 |
GLN | 0 |
GLY | 0 |
ILE | 0 |
LEU | 0 |
MET | 0 |
PHE | 0 |
PRO | 0 |
SER | 0 |
THR | 0 |
TRP | 0 |
TYR | 0 |
VAL | 0 |
ACE | 0 | Acetyl cap
NME | 0 | N-methyl amide cap
Non-canonical Amino Acids Recognized by QUELO¶
In addition to the 20 standard amino acids (and their charge variants) that are recognized by QUELO, as described in the previous section, QUELO also recognizes a variety of phosphorylated amino acids, which are sometimes found in protein structures. Phosphorylated amino acids that are recognized are shown in the table below. The PO4 group can exist as either PO4 (2-) or HPO4 (1-), which accounts for the two charge variations of each residue type. (For the phosphohistidines, the group is N-PO3 or N-HPO3). You must ensure you use the naming convention shown:
Residue Name | Charge State | Parent amino acid | Notes
PTR | -2 | TYR | Phosphotyrosine
Y1P | -1 | TYR | Phosphotyrosine
SEP | -2 | SER | Phosphoserine
S1P | -1 | SER | Phosphoserine
TPO | -2 | THR | Phosphothreonine
T1P | -1 | THR | Phosphothreonine
H1D | 0 | HIP (HIS) | Phosphohistidine, -HPO3 on ND
H2D | -1 | HIP (HIS) | Phosphohistidine, -PO3 on ND
H1E | 0 | HIP (HIS) | Phosphohistidine, -HPO3 on NE
H2E | -1 | HIP (HIS) | Phosphohistidine, -PO3 on NE
RNA Bases Recognized by QUELO¶
QUELO supports RNA bases, expecting the following residue names:
Residue Name | Notes
A | Adenine
C | Cytosine
G | Guanine
U | Uracil
A5 | Adenine at the 5' terminus without a phosphate group
C5 | Cytosine at the 5' terminus without a phosphate group
G5 | Guanine at the 5' terminus without a phosphate group
U5 | Uracil at the 5' terminus without a phosphate group
A3 | Adenine at the 3' terminus
C3 | Cytosine at the 3' terminus
G3 | Guanine at the 3' terminus
U3 | Uracil at the 3' terminus
AMP | Adenine at the 5' terminus with a singly protonated phosphate group
GMP | Cytosine at the 5' terminus with a singly protonated phosphate group
CMP | Guanine at the 5' terminus with a singly protonated phosphate group
UMP | Uracil at the 5' terminus with a singly protonated phosphate group
Reserved Ligand Residue Name¶
QUELO reserves the residue name UNL for the ligand residue. The user should not use this residue name in the input PDB file.
Supported Atom Types¶
At present, QUELO is designed to deal with all types of standard organic molecules, but there are limitations on what metal atoms and ions can be included:
Organic molecules: All standard organic atom types that are found in proteins, ligands, and cofactors are supported (H, C, N, O, P, S, Cl, Br, F, I, etc.)
Metal atoms: QUELO supports all metals that appear in the Protein Data Bank at least a few times. The atom name of the metal must match the residue name. Note that iron and copper have more than one possible charge state; pay particular care when choosing their residue/atom names and element names. Here is a list of the supported metals, alongside their required names and charges:
Residue/Atom Name | Charge State | Element Name
ZN | +2 | Zn2+
Na+ | +1 | Na
MG | +2 | Mg2+
FE2 | +2 | Fe2+
FE | +3 | Fe3+
CA | +2 | Ca2+
CU1 | +1 | Cu+
CU | +2 | Cu2+
CO | +2 | Co2+
NI | +2 | Ni2+
MN | +2 | Mn2+
K | +1 | K
V2+ | +2 | V2+
CD | +2 | Cd2+
HG | +2 | Hg2+
Solvent ions: QUELO reads in Na+ and Cl- ions from the input PDB file. Additional Na+ and Cl- ions will also be generated to match physiological concentration and neutralize the simulated system.
The Na+ residue must have the residue name Na+ to be recognized.
The Cl- residue must have the residue name Cl- to be recognized.Waters: The program supports water molecules with either of the following residue names: WAT or HOH.
RNA bases: QUELO support RNA bases, assuming their names conform to the Amber naming scheme.
Rules for Specifying Cofactors¶
Cofactors are a special class of molecules when it comes to processing a PDB file. Residue names are matched to identify proteins, certain ions (Na+, Cl-), waters (HOH or WAT), and metal atoms (ZN, MG, FE2, FE, CA, CU1, CU, CO, NI, MN, K, V2+, CD, HG). Any residues not otherwise recognized are termed “cofactors.”
The user is responsible for cleaning up the input PDB file before uploading it into the platform. Details of what this means are described earlier in this chapter. One task for the user is to remove any cofactors that should not be included for the simulation.
If the user has decided that certain cofactors are important to include in the simulation, then the following
rules must be adhered to to ensure that the cofactor molecules are properly processed by the QUELO platform.
Contiguous atoms sharing the same residue number and chain are considered part of the same cofactor
Note that this means that if the cofactor initially consists of multiple residues, both the residue names and residue numbers must be modified so that all atoms are part of the same residue
Cofactors cannot have a name that is the same as those of standard recognized residues (standard amino acids, etc.)
Cofactors cannot contain atoms with the names C, N, CA, or O. These names are reserved for amino acids. Inclusion of atoms with these names in a cofactor will raise an error.
Cofactors must have reasonable geometries and must have all atoms (including hydrogens) present.
There should be CONECT records within the PDB file that define the topology (bonds) for each cofactor, including bond orders. For example, if atom 1 has a double bond with atom 2 and single bonds with atoms 3 and 4, the expected CONECT line is:
CONECT 1 2 2 3 4
If CONECT records are not provided, the cofactor is rejected.
Atoms assigned a formal charge in the molecule need to be identified with special atom types in fields 77-80 (see PDB file fields in the previous section). Two special atom types are recognized in this context:
“O1-”: Charged oxygen in carboxylates, etc.
“N1+”: Charged nitrogen in amines, etc.
The atomic charges are still determined via fitting of the results from quantum mechanics for atoms specified as having a formal charge. These designations merely ensure the proper distribution.
QM Region Selection¶
Running a QM/MM simulation requires the specification of the set of atoms that will be treated using a quantum mechanical representation. The selected atoms are treated using QM, and the remaining atoms in the system are treated using a classical molecular mechanics (MM) force field.
Significant increases in the size of the QM region will necessarily increase the computing cost and lengthen the turnaround time for a calculation. For this reason, one generally identifies a QM region just large enough to contain the most significant interactions of the binding region with the bound ligand. QM regions of 100-200 or fewer atoms tend to reflect a good compromise, although one can increase the QM region to up to 300 or so atoms and still obtain turnaround on a timescale that is acceptable by modern drug discovery standards.
Atoms are selected on a full residue basis, with the exception of certain backbone atoms on adjoining protein residues, which can be included to improve stability.
QM Region Specification¶
The QM region can either be generated automatically, or else the user can define the region by specifying exactly the residue set they wish to be included in this region. No matter which option is chosen, the mutating ligand must always be included in the QM region.
In the case of auto-generated QM region residues, a set of criteria are applied:
Cutoff distance: The cutoff distance is fixed at 5 Angstroms and cannot be modified by the user. The cutoff distance is used to automatically identify residues that are eligible for further consideration as part of the QM region. The cutoff distance is not used if the user is specifying the QM region manually, via residue definitions in the text input box (below). A search is performed to identify residues for which at least one atom is <= the cutoff distance from at least one atom of the mutating ligand. Any residue that meets this search criterion will pass the cutoff selection filter. Selection is performed on a full-residue basis (if one or more atoms of the residue are close enough, the entire residue is included). The cutoff distance is applied to both the protein receptor and any cofactors that may have been included in the input PDB file. Water molecules in the input PDB file are not included in the cutoff.
Region max atoms: The maximum number of atoms that will be included in the QM region when auto-selection is being carried out. If the number of atoms selected for auto-select is greater than region max atoms, the selection will be trimmed (on a residue basis) until the region max atoms criterion is met.
Note that if you are in Expert Mode, there is an atom maximum threshold, which is applied only to mutations between residues that have differing net formal charges. The QM region for mutations where a formal charge change occurs is selected via a different approach than the remainder of the mutations–one based on assessing the polarizability of the binding region. Typically, a somewhat larger maximum number of atoms is required for these mutations. A default value of 130 is applied, but if you are in Expert mode, you can change this. This option appears below the QM Region Summary
Selection algorithm=Simple: Filtering to define the QM region is first performed based on the cutoff distance. If the number of atoms in the filtered group is greater than the region max atoms, the minimum distance of each residue in the filtered region to the mutating ligand will be determined, and the list will be sorted from largest to smallest. Residues will be removed from the list in order, starting with the largest minimum distance, until the region max atoms criterion is met.
Selection algorithm=Optimal: Residues that are within the cutoff distance are evaluated for whether they are making specific high-value interactions with the ligand. A Significance Index, based on whether it is making specific high-value interactions, with be determined for each residue. Residues are removed from the list of residues within the cutoff distance in reverse order of Significance Index (least_significant -> most_significant) until the region max atoms filter is met. The Significance Index for each residue is determined by a combination of
Distance to the mutating ligand (closer is better)
Reduced significance for residues that present only aliphatic or non-polar sidechains to the mutating ligand
If two (non-ligand) residues in the Optimal set are also involved in a salt bridge with one another, both residues will be given the same significance index (the greater of the values for the two residues)
The Residue Definition Text Box: In the middle of this section, you will find a text box. In this box, the definitions of the residues that will be included in the QM region are provided. As noted, you must always include the ligand in the QM region definition. To do this, use the keyword “ligand” (all lowercase and no quotes).
The Residue Definition Text Box can be populated either automatically (using the “Suggest a QM Region” button (below)), or else the user can provide a series of residue definitions “by hand”.
The user can specify the following residue types to be part of the QM region:
Protein residues (amino acids, capping groups)
Cofactors
Waters (HOH or WAT) when acting as ligands of metal ions
The syntax for specifying atoms in the SELECT mode is:
RES_NAME:RES_CHAIN:RES_NUMBER-TYPEThere is a single exception to the above syntax, and that is the specification for the interacting ligand, which uses the keyword ligand, alone on a line.
Examples:
ligandPHE:A:42-allTYR: :109-sidechainRES_NAME: 3-character residue name. The name must exactly match the name in the input PDB. Case is ignored.
RES_CHAIN: Chain identifier that exactly matches that in the input PDB file. If there is no chain ID in the input file, leave this blank. Case is ignored.
RES_NUMBER: The residue number. It must exactly match the residue number in the input PDB file.
TYPE
all: Include all atoms in the residue (this is the only allowable option for cofactors and for non-standard amino acids).
backbone: Include only backbone atoms (C, O, CA, HA, N, H)
sidechain: Include only sidechain atoms (everything but the backbone)
backboneC: Include only the backbone atoms (C, O, CA, HA). Using this TYPE option requires that this is not a terminal residue, that the next residue in the protein backbone is also included in the SELECT set, and that the following residue is specified with TYPE “all” or “backbone”
backboneN: Include only the backbone atoms (CA, HA, N, H). Using this TYPE option requires that this is not a terminal residue, that the preceding residue in the protein backbone is also included in the SELECT set, and that the preceding residue is specified with TYPE “all” or “backbone”
Suggest a QM Region: This button will auto-populate the Residue Definition Text Box, based on the criteria (Cutoff distance, Region max atoms, Selection algorithm) described above. After clicking this button, the Residue Definition Text Box will contain a series of residue identifiers. The user is free to modify the auto-generated list, removing or adding to the list, as desired.
Validate: Validation reads the list of residue specifiers in the Residue Definition Text Box and ensures that the definitions are valid for the input PDB file. Validation is automatically performed if the text box is filled using the Suggest a QM Region button. The Validate button is only active when new or changed definitions are included in the Residue Definition Text Box that have not already been validated. Functions of Validation, which must be carried out before running the simulation, include:
Ensure every residue definition matches a residue in the input PDB file
Add capping groups, if necessary, to ensure usable QM/MM boundaries
Determine the total number of atoms specified by the residues included in the QM region
A user-supplied or modified definition of the QM region (i.e. one not generated using the Suggest a QM Region button) will not be subject to the Maximum Atoms filter. If you modify the region, after validation, be sure to note the total number of atoms in the QM region (QM Region Summary, below) and make sure it is reasonable.
Visualize: Creates a pop-up window that shows the selected QM-region residues in the context of the protein/ligand complex. The reference ligand will be shown in blue, bound to the protein. The protein residues included in the QM region will be shown in red. You can rotate the view using your mouse (left button) or translate the view with the mouse (right button). Double-click on any atom to reset that atom as the center for rotations. If you hover the mouse over any atom, information on the atom and the associated residue (name, number) will pop up. To close the pop-up window, click the “X” near the top right of the box.
QM Region Summary: To the right of this section, you will find the QM Region Summary, which is populated after you validate the QM region contents. (The definition is auto-validated, and this region is auto-populated if you use the Suggest a QM Region button). The header is labeled with “Charge Conserving” or “Charge Changing” based on the mutations considered. In this section you will find:
Status: Whether the contents of the definition box properly validated. The possibilities are “Validated” or “Invalid.” If the contents are Invalid (there is a problem) or if they are valid but a warning was issued, the status will be shown in red, and you can ascertain more information by hovering the cursor over the question mark.
Number of link atoms: Number of hydrogen atoms that were added to address unfulfilled valencies that arose from using separated protein residues. These are required for the QM simulation.
Number of total QM atoms: Total atoms in the QM region, excluding the ligand itself. Based on the reference ligand.
Total QM charges: Total charge of the QM region (ligand + other residues), based on the reference ligand.
Selection Syntax for RNA Bases¶
When the system contains RNA and the user wishes to include some bases in the quantum region, they need to respect corresponding selection syntax. Instead of “sidechain” and “backbone”, the available selection types are:
base: Include all atoms in the nucleotide base.sugar: Include all atoms in the nucleotide sugar. Note that this does not include one oxygen (PDB coventional name O3’) which should instead be included in the QM region alongside the following phosphate.phosphate: Include all atoms in the nucleotide phosphate. Note that this is missing one oxygen (PDB coventional name O3’) which is considered part of the preceding nucleotide residue.5cap: Include phosphate oxygen (PDB coventional name O3’) matching the following nucleotide phosphate. This cap is required if the following residue phosphate is included.all: Include all atoms on the nucleotide.
The residue specification works the same as for proteins. Here are some examples:
G:A:43-base
U:C:2-sugar
A:H:105-all
Several selection types (base, sugar, phosphate, and 5cap) can be simultaneously specified for one residue if desired. The instructions are written as one word and the order does not matter. Here are some examples:
G:A:43-basesugar
U:C:2-sugarphosphate
A:B:2-sugarbasephosphate5cap
Validation will automatically apply 5cap selections when necessary to cap phosphates included in the QM region. Here is an example of a validated QM region:
C:A:10-5cap
U:A:11-phosphate
The QUELO FEP Command Line Interface (CLI)¶
The CLI vs the GUI¶
The QUELO FEP Command Line Interface (CLI) is an alternative way to access the functionality of this platform. It provides the same functionality as the GUI version that is described in previously chapters, and it is not required that you either install or use the CLI. Whether you use GUI or CLI access is entirely a matter of preference. Note that the CLI requires you install local software in a Python environment, and so it is limited to platforms where you have ready Linux access, and where you can install some requisite software.
In contrast, the GUI is accessed through a standard browser and requires no software installation. Therefore, GUI access is available on a much larger array of devices (computers, tablets, phones, etc.).
Note that both CLI and GUI initiated calculations are stored on the backend in the same databases. That means you can access calculations that were initially run from the CLI through the GUI, and vice-versa.
Installation Note¶
Note that before you can run the CLI, you need to have installed the CLI on your computer, following the instructions provided in the chapter The QUELO Command Line Interface (CLI): Installation. Installation only needs to be performed once (unless you need to update the software). If you want to use the CLI, you must install the software first, using the step-by-step instructions in that chapter.
Assuming you have followed the instructions in the installation chapter and installed the QUELO CLI into a virtual environment under Miniconda, each time you log into the computer and want to access the CLI, you will need to activate the appropriate virtual environment. This is done using the following command:
(base) prompt: conda activate QUELO
Overview¶
The previous chapters have described the functionality of the QUELO platform, and how to access the platform through the browser-based GUI. As noted, an identical set of functionality is accessible through the CLI.
The CLI runs in the Linux environment. To run the CLI, some infrastructure must be installed using a small number of simple commands, as are described in the Installation section (next chapter). Once the CLI is installed, the user can invoke it using the sbb command. The available sbb commands and syntax are described in the sections below.
For details on implementation of the QUELO process, the user is referred to previous chapters.
In the sections that follow, the following should be noted:
Enclosing arguments with double quotes is required for arguments with space or other special characters, e.g. a batch name with embedded spaces.
Optional flags are enclosed in square braces.
Flags that require arguments are followed by a value in angle braces.
Running the QSimulate CLI¶
Once you have installed the CLI package, following the instructions in the Installation chapter, and you have activated the appropriate virtual environment, using the command above, you will be able to run calculations from the command line. The software is invoked using the “sbb” command, followed by options and keywords that describe exactly what you want to do. For example, sbb -h will return a top-level help menu:
(QUELO) Prompt:~/Desktop$ sbb -h
usage: sbb [-h] [-V] [--rc RC] Commands ...
CLI interface for the QSimulate Platform
positional arguments:
Commands
session Logging in, logging out, setting the server address
batch Create a batch job and set options for the job, run or delete a batch
molecule Attach molecule file(s) to a created batch
options:
-h/--help Show this help message and exit
-V/--version Show version and exit
--rc RC Set an alternate config file
Session Command¶
sbb session
[set-url --url <URL_TO_ACCESS_QSIMULATE>]
[login -u/--user <USERNAME> -p/--password <PASSWORD> -i/--key KEY]
[logout]
[update-cert]
Session Set-URL¶
Logging into the CLI requires that you fist specify the name of the server you will be using for your calculations, and then actually logging into that server.
To specify the URL of the server (a URL you will have been provided by QSimulate or your system administrator) use the following command. This command only needs to be performed once during a session, even if you log out and back in again during that session.
sbb session set-url --url URL_TO_ACCESS_QSIMULATE
[For example: sbb session set-url --url my-company.qsimulate.com]
Session Login and Logout¶
To log into the server, you use the command
sbb session login -u/--user USERNAME -p/--password PASSWORD -i/--key KEY
USERNAME and PASSWORD are replaced by the credentials you set up for the QSimulate platform. If you do not wish to write your password in plaintext in the command line, you can omit the “-p PASSWORD” part of the command, in which case the server will prompt you to enter your password.
There is also an option to log in using a key. The easiest way of doing this is using sbb key. For example, after logging in, you can do the following:
sbb key create -i qspkey > ~/.ssh/mykey
This will generate a private key and store it in .ssh/mykey, as well as storing the public key on the server. qspkey refers to the name of the key for the purposes of listing and/or removing by QUELO CLI, while .ssh/mykey is the file on your file system. The next session, you can log in with the command:
sbb session login -u/--user USERNAME -i ~/.ssh/mykey
If you want to remove your key, you can do it by this command:
sbb key remove -i qspkey
If your login was successful, you will see the message
Login successful.
To subsequently logout of the server, use the command
sbb session logout
Note that you will be auto-logged off the server after 15 minutes of inactivity. If you attempt to issue a sbb command that requires a response from the server after you have been logged out, you’ll be shown the message
Please login
Session Update-Certificate¶
It is unlikely you’ll ever need to manually update the SSL certificate. But if you do, the certificate can be updated using the commmand
sbb session update-cert
Batch Command¶
Within the QSimulate platform, calculations you want to run are termed “batches”. Each calculation run is a “batch”. The batch command can be used to list and/or examine batch calculations you have already submitted, or to create and submit a new batch calculation. The main level options for batch command are:
sbb batch [create]
[create-from-file]
[run]
[create-and-run]
[list]
[results]
[delete]
[stop]
[restart]
[options]
[preprocess]
[clone]
[progress]
[update-options]
The standard workflow for setting up a QUELO QM/MM-FEP calculation requires eight commands:
sbb batch create
sbb molecule create (receptor)
sbb molecule create (reference ligand)
sbb molecule create (additional ligands)
sbb batch preprocess generate_mutation_map
sbb batch run (for QM/MM job, this will carry out all preprocessing up to QM region selection, then pause)
sbb batch preprocess suggest_qm_region
sbb batch resume (resumes the job after selecting the QM region).
The first sets the options for the calculation. Commands 2-4 attach molecular input files. The fifth command determines the closed perturbation map. The sixth establishes the quantum region (if performing a QM/MM-FEP calculation). The seventh submits the calculation for execution.
The standard workflow for setting up a QUELO MM-FEP calculation requires six commands:
sbb batch create
sbb molecule create (receptor)
sbb molecule create (reference ligand)
sbb molecule create (additional ligands)
sbb batch preprocess generate_mutation_map
sbb batch run
Important considerations for scripted execution¶
If you plan to run the CLI commands from a script or pipe, it is crucial to understand that while certain commands (molecule create, batch preprocess) will immediately return the user to the command line, the actions performed by these commands (especially batch preprocess) can take some time. If you attempt to submit (run) a job for which the intermediate steps have not completed, you will get an error message. To circumvent the issue, you will want to to insert code in your submission workflow after each of these commands to ensure the script pauses until the previous command has completed.
The various commands are detailed in the sections below
Batch Create¶
sbb batch create \
-b/--batch <batch_name> \
-c qsp_fep {qsp_fep, qsp_fep_gcmc}\
[--dry-run ]
[--quelo_config {ai,classical,xtb,xtb2}]
[--fep_timestep FEP_TIMESTEP]
[--seed SEED]
[--eq_runtime EQ_RUNTIME]
[--heat_runtime HEAT_RUNTIME]
[--pres_runtime PRES_RUNTIME]
[--qm_eq_runtime QM_EQ_RUNTIME]
[--charge_method {resp,bcc,mul,none}]
[--fep_num_lambda_points FEP_NUM_LAMBDA_POINTS]
[--fep_eq_runtime FEP_EQ_RUNTIME]
[--fep_runtime FEP_RUNTIME]
[--calc_solv_energy]
[--no-calc_solv_energy]
Calculation options for a batch are configured when this command is called. After creating the batch, you will also need to attach input molecule files (see “sbb molecule”), create a perturbation map (see “sbb batch preprocess generate_mutation_map”), and assign the QM region, if any (see “sbb batch preprocess suggest_qm_region”). The job cannot be run until these additional steps are taken. See below.
Sbb batch create options detail is provided below. Additional explanation for some of these options can be found in the preceeding chapters that detail the GUI panel.
-b/--batch <batch_name>
Required. Defines the name of a new batch name to be created.
-c qsp_fep {qsp_fep,qsp_fep_gcmc}
Required. Indicates that you are setting up a QUELO FEP calculation. An option must be specified with this flag. “qsp_fep” sets up a standard FEP simulation. “qsp_fep_gcmc” sets up a Grand Canonical Monte Carlo (GCMC) FEP simulation, where insertion of buried water molecules will be periodically attempted during the simulation.
--dry-run
If the dry-run argument is specified, the calculation options will be written to standard out in JSON format and no batch calculation will be created.
--quelo_config {ai,classical,xtb,xtb2}
Defines the type of free energy perturbation: AI, classical, xtb or xtb2.
--fep_timestep FEP_TIMESTEP
(In picoseconds). Default = 0.002. The integration timestep used during molecular dynamics. One Generally should not exceed 0.002ps with SHAKE bond constraints on (default), or 0.0005ps with SHAKE off.
--seed SEED
Defines a random number seed (integer) that is used to create a pseudo-random distribution of values for initial velocity assignments.
--charge_method {resp,bcc,mul,none}
Method used to calculate the partial charges on atoms for the MM interactions. Options are RESP (default), BCC, MUL (Mulliken) or none (charges set to 0).
--eq_runtime EQ_RUNTIME
In nanoseconds. Default = 0.25. The total amount of MD equilibration to perform before starting the FEP runs. This value does not include any MD requested by Heating and Pressurization (below). This MD equilibration is performed with an MM representation of the entire system. (For QM/MM systems, additional equilibration will subsequently be performed). MD is performed using the lambda=0.5 system.
--heat_runtime HEAT_RUNTIME
In nanoseconds. Default = 0.10. The temperature is ramped up from an initial temperature of 0K to the target temperature over the specified number of nanoseconds of simulation.
--pres_runtime PRES_RUNTIME
In nanoseconds. Default is 0.10. The pressure is ramped up from the value calculated from the input system and velocities to the target pressure over the specified number of nanoseconds of simulation.
--qm_eq_runtime QM_EQ_RUNTIME
In nanoseconds. Default = 1.00. The total amount of MD equilibration to perform using the QM/MM energy function before starting the FEP runs. This MD equilibration is performed starting with the output of the MM-based equilibration (above). MD is performed using the lambda=0.5 system. This option is ignored if no QM region is defined.
--fep_num_lambda_points FEP_NUM_LAMBDA_POINTS
Default = 13 (MM) or 14 (QM/MM) or 16 (AI). Number of lambda windows (or points) used for the FEP calculation. For MM, all lambda windows are equally spaced. The lambda windows are denser at the beginning and end (around lambda of 0 and 1) for computational efficiency.
--fep_eq_runtime FEP_EQ_RUNTIME
In nanoseconds. Default is 0.5. The number of nanoseconds of MD equilibration to perform at each lambda window before data collection starts.
--fep_runtime FEP_RUNTIME
In nanoseconds. Default is 5.0 (MM) or 2.0 (QM/MM). The amount of molecular dynamics sampling to perform for each lambda window to evaluate the free energy difference across that window.
--calc_solv_energy
--no-calc_solv_energy (D)
Option to request that the relative solvation free energy for each pair of ligands be determined. The relative binding free energy of each pair of ligands is always calculated.
Batch Create-From-File¶
sbb batch create-from-file \
-b/--batch <batch_name> \
-d input_data_file
You should not specify this option. This option is reserved for developer use, or if QSimualte is working with an end user to track down anomalous behavior.
Batch Run¶
sbb batch run \
-b/--batch <batch_name>
[--fleet-type {ondemand,spot}]
[--priority <priority>]
This command will submit (run) a batch job that was previously created, to which molecule files have been attached, and which has the status of “staged”. Note that before you can run a job, you must associate three molecule input files to the job using the Molecule Create command (see below), and you need to have generated the perturbation map and (for QM/MM jobs) the QM region using the Batch Preprocess commands. If you attempt to run a batch for which you have not defined this additional input, the calculation will not run.
If you attempt to issue the batch run command before the molecule uploads or Batch Preprocess commands have fully completed, (i.e. a map is still in the process of being generated), you will receive an error message.
--fleet-type {ondemand,spot}
By default, QUELO calculations are run using “spot” instances on the AWS cloud, which typically allows for far better pricing, but which can be pre-empted by AWS at times of heavy use. The QSimulate platform efficiently handles spot interruptions by periodically saving drop files and automatically restarting any jobs that get interrupted. The user will not be aware of these. On rare occasions, the usage at AWS may be so high that the rate of spot instance interrupts is high enough to significantly slow the time to job completion. In such a case, one could use the on-demand option to instead run using the significantly more expensive non-interruptable job queue. However, this option is generally not needed or recommended.
--priority <priority>
Default is 30.
The queueing priority of the jobs, relative to other jobs the same user/company has submitted. The priority can be used for ensuring a particular job gets sent for execution before others in the user’s queue. It has no affect at all on performance/turnaround once the job exits the queue and starts executing. The value can be between 10 and 99, and lower values result in higher priority. The default value for GUI jobs is 50, so you can also use this value to change the relative execution weight of CLI vs GUI jobs.
Batch List¶
sbb batch list [-o/--output-style {table/csv/html/json}]
Returns a list of all batch calculations that have been created and/or run. All batch calculations run from the account will be shown, including calculations (if any) performed using the GUI interface.
Four columns are presented in the list. For QUELO FEP jobs, the “Smiles” column is not applicable. (The “Smiles” column is used for other calculation types (QuantumFP)).
The optional output-style specifier will designate the format of the list. The default is “table”. An example output table would be:
+------------------------+-----------+--------------------+----------+
| Batch Name | Molecules | Type | Status |
+------------------------+-----------+--------------------+----------+
| First Run | - | qsp_fep | complete |
| Run number 2 | - | qsp_fep | staged |
+------------------------+-----------+--------------------+----------+
The same output in csv format (sbb batch list -o csv) would be:
Batch Name,Molecules,Type,Status
First Run,-,qsp_fep,complete
Run number 2,-,qsp_fep,staged
html format is intended for use in Jupyter notebooks.
Potential status values include: staged, running, complete, stopped, and failed.
Staged: Job has been created but not submitted to run. Or a previously stopped job in the process of resuming.
Running: Job is currently in process on the servers
Complete: Job has completed successfully
Stopped: Job was paused using the batch stop command
Failed: Job failed to complete successfully
Batch Results¶
sbb batch results \
-b/--batch <batch_name> \
[-o/--output-style {table/csv/html/json}]
[--after <TIMESTAMP>]
[--before <TIMESTAMP>]
[-d/--dir <OUTPUT_DIRECTORY>]
This command will return the results for the completed batch calculation with the name batch_name. If batch_name includes embedded spaces, you must enclose the full name with double quotes. The result is streamed to standard out, and you will typically redirect to a file. For example:
sbb batch results -b "Test Batch 01" --output-style csv > batch01.csv
The -o/-output-style format operates exactly as described above for Batch List.
--after <TIMESTAMP>
--before <TIMESTAMP>
Will return all batch jobs after, or before, the time specified. Standard date/time formats are accepted for TIMESTAMP, including the following. Note that the TIMESTAMP string should be enclosed in quotes.
'11 Mar 2023'
'11 Mar 2023 05:00:00'
'3/11/2023'
'2023-03-11 5am'
-d/--dir <OUTPUT_DIRECTORY>
By default, the output results are written to the current local directory, with the name specified by “batch_name”. If you specify this option, then the file will be written the OUTPUT_DIRECTORY given. OUTPUT_DIRECTORY should be surrounded by quote marks.
Batch Delete¶
sbb batch delete -b/--batch <batch_name>
This command will delete the named batch. Note that the delete will be performed immediately and there is no subsequent user verification, so be careful when using this command.
Batch Stop¶
sbb batch stop -b/--batch <batch_name>
This command will stop the named batch. The state of the calculation and all intermediate results are retained when the calculation is stopped, and it can subsequently be restarted using the batch restart command (below). This command can only be successfully be executed for a job whose current status is “Running”.
Batch Restart¶
sbb batch restart \
-b/--batch <batch_name>
[--fleet-type {spot,ondemaand}]
This command will restart a previously stopped batch. This command can only be successfully be executed for a job whose current status is “Stopped”. The fleet-type option is described under Batch Run. Typically, you would not change this from the default of “spot”.
Batch Options¶
sbb batch options -b/--batch <batch_name>
This command will report the options used for the named batch in JSON format. The report is sent to standard output.
For example, a QM/MM-FEP job originally run from the GUI with all default options would return this JSON report:
{"jobType": "qsp_fep",
"type": "custom",
"options": {
"qm_method": "xtb",
"qm_region_max_atoms": 200,
"qm_chop_method": "hotspots",
"qm_region_charge_max_atoms": 40,
"charge_qm_chop_method": "hotspots",
"fep_timestep": 0.002,
"seed": -1,
"eq_runtime": 0.25,
"heat_runtime": 0.1,
"pres_runtime": 0.1,
"qm_eq_runtime": 1,
"fep_num_lambda_points": 14,
"fep_eq_runtime": 0.5,
"fep_runtime": 2,
"calc_solv_energy": false}
}
}
Batch Preprocess¶
sbb batch preprocess
{generate_mutation_map
upload_mutation_map
show_mutation_map
check_mutation_map
suggest_qm_region
upload_qm_region
show_qm_region}
-b/--batch <batch_name>
The batch preprocess command is required to complete set up of a QUELO calculation.
Pre-processing accomplishes two things:
Establish the perturbation map
Establish what atoms will be included in the quantum region (for QM/MM calculations)
When you issue the sbb batch preprocess command you must specify one of the options shown (described below), as well as the name of the batch calculation to which the command is applied.
Command options (choose one):
Batch Preprocess¶
sbb batch preprocess generate_mutation_map
[--charge_mutation_max_num <VALUE>]
[--scoring_method {3D,2D,PERFECT}]
[--map_style {default,star_map}]
-b/--batch <batch_name>
Generates a perturbation map that connects all the supplied ligands. Each connector (edge) of the map represents a perturbation (mutation) that will be performed. The map is created to optimize maximum common substructure overlap among the ligands.
–charge_mutation_max_num <VALUE> (Default = 2). Defines the maximum number of connections between ligand nodes in the perturbation map that have differing formal charges.
–scoring_method: The method to align ligands and score the alignments. 3D and 2D alignments are available, as well as assuming a perfectly prealigned set of ligands (PERFECT).
–map_style: The style of the perturbation map to be used. The default option refers to a map with redundant perturbations to correct for hysteresis while there is also the option (star_map) to create a simple “star” map, with a single mutation connecting the reference ligand to each of the other input ligands. A star map reduces the number of calculated mutations and cost of the calculation, but the results may be somewhat less accurate. Default is no star map.
sbb batch preprocess upload_mutation_map
--mutation_map <MUTATION_MAP>
-b/--batch <batch_name>
Allows the user to upload a perturbation map to be used. To function properly, every ligand (node) of the map must be connected to the overall map by at least one edge (mutation). The user is responsible for ensuring that a valid map is uploaded. The map name and path is specified using the –mutation_map option, which is required. The format of the map file is a series of two strings per line, separated by a comma. The strings are the names of the two ligands to be connected by a mutation (edge). For example:
17,21
17,test1
test1,21
would specify a map for three ligands (named, “17”,”21”, and “test1”) connected together as a triangle.
sbb batch preprocess show_mutation_map
-b/--batch <batch_name>
Downloads the perturbation map for the specified batch job. For example, for the triangular that connects ligand nodes 17, 21, and “test2”, the result of this command would appear as:
Perturbation map:
17,21
17,test1
test1,21
sbb batch preprocess check_mutation_map
-b/--batch <batch_name>
This command checks the status of perturbation map generation. If the perturbation map is still in progress, the command will return a message showing the percent completeness, e.g.:
Perturbation map generation progress: 18% complete
Once the perturbation map generation is complete, the command will return the message:
No perturbation map generation in progress.
Note that the above message will also appear immediately after you submit a generate_mutation_map command–before map generation has had a chance to start. So if you are implementing this command in a script to create a status-based pause, you need to ensure you give the command sufficient time to start before polling.
sbb batch preprocess suggest_qm_region
[--qm_region_max_atoms <QM_REGION_MAX_ATOMS>]
[--qm_chop_method {simple,hotspots}]
[--qm_region_charge_max_atoms <QM_REGION_CHARGE_MAX_ATOMS>]
-b/--batch <batch_name>
This command is used to request that the platform automatically choose a set of residues that will comprise the QM region for a QM/MM calculation. It is ignored for MM-only FEP simulations.
–qm_region_max_atoms <QM_REGION_MAX_ATOMS>: Default = 200. Maximum number of atoms that can be part of the QM region. This value is used when an automated approach to defining the QM region is used. This value is ignored if the user uploads a QM region definition using the “batch preprocess upload_qm_region” command (below). Note that the default value is very large–larger than often necessary, and using the default can result in calculations that are very expensive to run. It is generally recommended you adjust this value to a lower number. This value applies to mutations where the formal net charge of the ligand remains constant for the mutation. In cases where for the formal net charge of the ligand changes, the value given by “qm_region_charge_max_atoms” (below) is used instead.
–qm_chop_method {simple,hotspots (D)}: Default = hotspots. When the program suggests the atoms to include in the QM region, either a simple distance-based approach can be applied (simple), or a more sophisticated method can be used that prioritizes important interactions with the ligand (hotspots).
–qm_region_charge_max_atoms <QM_REGION_CHARGE_MAX_ATOMS>: Default = 130. Maximum number of atoms that can be part of the QM region for mutations where the formal charge of the ligand changes. This value is used when an automated approach to defining the QM region is used. This value is ignored if the user uploads a QM region definition using the “batch preprocess upload_qm_region” command (below). This value applies to mutations where the formal net charge of the ligand changes for the mutation. In cases where for the formal net charge of the ligand remainns constant, the value given by “qm_region_max_atoms” (above, see “sbb batch create”) is used instead.
sbb batch preprocess upload_qm_region
[--qm_region_instructions <QM_REGION_INSTRUCTIONS>]
[--charge_qm_region_instructions <CHARGE_QM_REGION_INSTRUCTIONS>]
-b/--batch <batch_name>
This command allows the user to upload a series of command that define the quantum region. If used, this command will overwrite any previously auto-generated quantum region. Two instructions sets can be uploaded, one for mutations where the formal charge of the ligand does not change, and one for the case where the formal charge of the ligand is changing. If you use this command, you must specify at least one of these files.
The format of the instruction sets is described in the chapter, “QM Region Selection.”
–qm_region_instructions <QM_REGION_INSTRUCTIONS>: Specifies a file that contains the commands to define the qm region atoms for mutations where the formal charge of the ligand does not change.
–charge_qm_region_instructions <QM_REGION_INSTRUCTIONS>: Specifies a file that contains the commands to define the qm region atoms for mutations where the formal charge of the ligand changes.
sbb batch preprocess show_qm_region
-b/--batch <batch_name>
This command will write to standard output information about the QM region definition for the specified batch calculation, as well some simple metrics.
Batch Clone¶
sbb batch clone
-b/--batch <batch_name>
-n NAME
This command will copy (“clone”) the entirety of the setup (options, map, QM region) for an existing batch job to a new batch job with a specified (new) name. The name for the cloned batch entry, NAME, must not be the same as an existing batch entry. Once a job is cloned, the clone exists as a separate and editable entity in the batch table.
Batch Progress¶
sbb batch progress
-b/--batch <batch_name>
This command will return progress metrics on the named batch calculation. For example, for a completed job:
OverallProgress
Parametrization: 100%
Equilibration: 100%
Production: 100%
Analysis: 100%
Before a job starts, all the metrics will be at 0%.
Batch Update-Options¶
sbb batch update-options \
-b/--batch <batch_name> \
[--quelo_config {ai,classical,xtb,xtb2}]
[--fep_timestep FEP_TIMESTEP]
[--seed SEED]
[--charge_method {resp,bcc,mul,none}]
[--eq_runtime EQ_RUNTIME]
[--heat_runtime HEAT_RUNTIME]
[--pres_runtime PRES_RUNTIME]
[--qm_eq_runtime QM_EQ_RUNTIME]
[--fep_num_lambda_points FEP_NUM_LAMBDA_POINTS]
[--fep_eq_runtime FEP_EQ_RUNTIME]
[--fep_runtime FEP_RUNTIME]
[--calc_solv_energy]
[--no-calc_solv_energy]
This command allows you to update any of the options set for the job when you created it (see “batch create”) or else cloned it (see “batch clone”). The list of options available is identical to those documented for “batch create” (except for the -c option, which cannot be modified once the job is created), and the user is referred to that section for more detail.
Molecule Command¶
The sbb molecule command is required, and allows you to attach molecules to the created batch job. It is required that you specify three molecule or molecule sets to run a FEP job:
The receptor molecule, specified as a PDB file
A reference ligand bound to the receptor (with coordinates that reflect the bound conformation)
A set of additional ligands for which the relative free energies of binding to the receptor will be calculated
sbb molecule [create]
[list]
[delete]
[describe]
Molecule Create¶
sbb molecule create
-b/--batch <batch_name>
-t/--type {sdf,mol2,pdb}
-f/--file <FILENAME>
-k/--kind {receptor,reference_ligand,additional_ligands}
The Molecule Create command is used to define the input molecules for the calculation. You will need to issue this command at least three times, to separately define the files containing the receptor, the reference ligand, and the additional ligands. Only one receptor or reference ligand can be specified, but more than one file containing additional ligands can be defined (all additional ligands in all files will be concatenated).
-b <batch_name>: Defines the name of the batch calculation to which these molecules will be attached. Name must already have been defined using a “batch create” or “batch clone” command.
-t {sdf,mol2,pdb}: Defines the format of the input file. The kind of file will limit the supported format. Attempting to use any other format will result in an error.
kind = receptor: Only PDB is supported
kind = reference_ligand: Only sdf or mol2 is supported
kind = additional_ligands: Only sdf or mol2 is supported
-f <FILENAME>: The name of the file that contains the molecule(s) being uploaded, including path if necessary.
-k {receptor,reference_ligand,additional_ligands}: Defines what type of input file is being specified.
receptor: (pdb format) The protein or nucleic acid receptor molecule. This file will also include any co-factors, structural waters, or other elements necessary for the simulation. Bulk solvent water and neutralizing counterions will be added to the system automatically, to fill a periodic box.
reference_ligand (sdf or mol2 format): A ligand that is bound to the receptor, with coordinates that reflect its binding conformation. Calculated free energies will be reported relative to this reference ligand.
additional_ligands (sdf or mol2 format): One or more additional ligands for which the relative free energies of binding will be determined. The coordinates of the molecules should be as bound to the receptor. You can define multiple sets of additional ligands by multiply issuing the molecule create command. All sets will be combined.
Molecule list¶
sbb molecule list
-b/--batch <batch_name>
[-o/--output-style {table (D),csv,html,json}]
[-k/--kind {receptor,reference_ligand,additional_ligands}]
Lists the molecules that have been uploaded to an existing batch with the name batch_name.
-b <batch_name>: Defines the name of the batch calculation to which these molecules will be attached. Name must already have been defined using a “batch create” or “batch clone” command.
-o {table,csv,html,json}: Define the format of the output file. Default is table.
-k {receptor,reference_ligand,additional_ligands}: Filter the output by the type of input. If this option is not specified, all molecule types will be listed.
Molecule Delete¶
sbb molecule delete
-b/--batch <batch_name>
-n/--name <NAME1 NAME2 ...>
Allows the user to delete specific molecules associated with the specified batch_name. To view a list of the names of molecules attached to a batch, use the Molecule List command (above). The names are in the left-most column, titled, “structure_label”.
-b <batch_name>: Defines the name of the batch calculation to which these molecules will be attached. Name must already have been defined using a “batch create” or “batch clone” command.
-n <NAME1 NAME2 …>: The names of one or more molecules that have been input for the system. Use the Molecule List command to see a list of names. You can specify as many names as desired with a single instance of the command. Separate names with white space. For example, to delete the molecules with names 21, RK17 and 4Q, issue this command:
sbb molecule delete -b Try -n 21 RK17 4Q
Note that after you delete molecules from the system, you must regenerate the perturbation map and quantum region using the Batch Preprocess command (above).
Molecule Describe¶
sbb molecule describe
-b/--batch <batch_name>
-n/--name <NAME>
Returns the structure file for the molecular entity with the given name. Molecule List will return a list of valid names.
-b <batch_name>: Defines the name of the batch calculation to which these molecules will be attached. Name must already have been defined using a “batch create” or “batch clone” command.
-n <NAME>: The names of one or more molecules that have been input for the system. Use the Molecule List command to see a list of names. Only one name can be specified. The structure will be presented in the same format it was input, i.e., the receptor will be returned as a PDB file and the ligands (including the reference ligand) will be returned as sdf files.
Specifying the Config File (RC)¶
By default, a configuration file is automatically generated for the user during the installation process, and is found in the home directory: $HOME/.sbb_cli_rc
There is generally no reason for the user to examine or change this file and the default is sufficient. In rare cases, you may wish to use a non-default RC file (although typically only if instructed to do so by your sysadmin or QSimulate). In this case, any sbb command you issue can be post-pended by the option
sbb -rc alternate_configuration_file
Which allows the user to specify an alternate RC file.
Example of running a calculation using the CLI¶
Set up environment and log in¶
In this example, we are going to assume you’ve set up the virtual environment, as described in the installation chapter, and called it QUELO. See the installation chapter for more details.
First, log into your Linux account. When you log in, you should be placed in the base, default Conda environment.
First, activate the QUELO virtual environment:
(base) Prompt$ conda activate QUELO
(QUELO) Prompt$
When you activate the virtual environment, the name of the environment will appear between parentheses at the beginning of your prompt. Once activated, the virtual environment stays activated for the remainder of your login session (or until you deactivate the session). The string shown for “Prompt” will depend on your account name and how your Linux machine was set up.
Log into your QSimulate CLI account.
(QUELO) Prompt$ sbb session set-url --url URL_TO_ACCESS_QSIMULATE
(QUELO) Prompt$ sbb session login --user YOUR_USER_NAME
Replace URL_TO_ACCESS_QSIMULATE with the URL provided by your system administrator or QSimulate. Replace YOUR_USER_NAME with the login name (or email) you use with the QSimulate account. You’ll be prompted to provide your password. If you are successful, you will see “Login successful.” on your screen.
Note that a login expires after 15 minutes of inactivity, and you’ll need to login again if that happens (using the same command).
Setting up the QUELO calculation¶
Important note:
It is important to note that the commands as described below should not be simply run from an unregulated command line script. Some of these commands take some time to execute–particularly the map generation and QM region determination. If you attempt to run subsequent commands before the work specified by previous commands has completed, the overall calculation will fail to submit.
In this example, we’ll go through a simple example of setting up a QM/MM-FEP simulation using QUELO. For simplicity, we’ll stick to all defaults, where available.
To run QUELO, you will need to have three files:
Receptor PDB file (receptor.pdb)
Reference ligand bound to receptor in SDF format (reference.sdf)
Additional ligands in SDF format. (additional.sdf)
While you can use any name you desire for these files, for the purposes of this tutorial, we are calling them, generically, “receptor.pdb”, “reference.sdf”, and “additional.sdf”, respectively.
At this point, you’ll want to create a new Batch calculation. You should first look at the names of any Batch calculations you have already run from this account, because you’ll need to assign a name, and it has to be unique. To look at the list of calculations you have already run, issue the command:
(QUELO) Prompt$ sbb batch list
This will show a table of all Batch calculations you have created, for example:
+------------------------+--------+--------------------+----------+
| Batch Name | Smiles | Type | Status |
+------------------------+--------+--------------------+----------+
| First Run | - | qsp_fep | complete |
| Run number 2 | - | qsp_fep | complete |
+------------------------+--------+--------------------+----------+
This table will include all Batch calculations you have created, whether from the CLI or the GUI. When creating a new Batch calculation, you need to choose a name not already in the table. (The CLI will let you know if you try to make a new Batch with the same name). For FEP simulations, the Smiles column is not used, and will contain a dash.
To create a new batch calculation named “my_new_batch” use the command:
(QUELO) Prompt$ sbb batch create \
-b my_new_batch \
-c qsp_fep \
--qm_method xtb \
--request_stop
This command will create a new Batch calculation that you can work on, named “my_new_batch”. Because we’ve chosen to accept all the defaults for the calculation, there are only a few flags we need to specify. In particular, we need to specify “-c qsp_fep” to indicate we’re running a FEP simulation. And we need to specify “xtb” for “–qm_method”, to get a QM/MM-FEP calculation (the default is an MM-FEP calculation with no quantum region). The “–request_stop” option is not technically required (it is the default), but it’s included here for clarity. This option will ensure that the batch calculation stops after all pre-processing up to the point where the QM region needs to be defined.
Next, we need to attach to this Batch calculation the input molecules that are required. We do this with a set of three invocations of the “sbb molecule command create” command:
(QUELO) Prompt$ sbb molecule create \
-b my_new_batch
-t pdb
-f receptor.pdb
-k receptor
(QUELO) Prompt$ sbb molecule create \
-b my_new_batch
-t sdf
-f reference.sdf
-k reference_ligand
(QUELO) Prompt$ sbb molecule create \
-b my_new_batch
-t sdf
-f additional.sdf
-k additional_ligands
These commands, as written, assumes the input files are in the directory you’re working from. If they aren’t, just add the correct path(s) before the name(s) of the file(s) in the command(s).
Now that we’ve attached the necessary files, we need to generate the perturbation map and define the residues that will be treated using quantum mechanics.
To generate the perturbation map automatically:
(QUELO) Prompt$ sbb batch preprocess generate_mutation_map -b my_new_batch
At this point, we still need to define the quantum region. Before that can be done, all other preprocessing needs to be carried out by the program. So we will start the batch, which will run until all preprocessing is complete and then pause. After the pause, we can define the QM region and continue.
- ::
(QUELO) Prompt$ sbb batch run
It may take several minutes for this portion of the setup to complete. You can monitor the status of the setup using the command “sbb batch list”. Wait until you see the status of your job listed as “stopped”, at which point you can proceed to define the quantum region.
To automatically assign the quantum region:
(QUELO) Prompt$ sbb batch preprocess suggest_qm_region --qm_region_max_atoms 80 -b my_new_batch
Note that we’re overriding the default value for the maximum size of the qm region. The default is 200 atoms, which is fairly large and can result in computationally expensive simulations. Here, we have reduce the maximum number of atoms to 80 using the “–qm_region_max_atoms” flag. The residues will be auto-chosen by the program based on “hotspot” interaction analysis.
Finally, we need to resume the Batch calculation we’ve added the QM definition to:
(QUELO) Prompt$ sbb batch resume -b my_new_batch
This will submit the Batch job to the CLI servers for execution.
Using the “sbb batch list” command, you can track the progress of the job until it completes. Eventually, you’ll see the table for batch list look something like:
+------------------------+--------+--------------------+----------+
| Batch Name | Smiles | Type | Status |
+------------------------+--------+--------------------+----------+
| First Run | - | qsp_fep | complete |
| Run number 2 | - | qsp_fep | complete |
| my_new_batch | - | qsp_fep | complete |
+------------------------+--------+--------------------+----------+
Once your job is complete, you can download the results. The following command will download a results table in CSV format:
(QUELO) Prompt$ sbb batch results -b my_new_batch -o csv > my_new_batch.csv
The results table will be in the file my_new_batch.csv. You can view this file using a text editor, or import it into a spreadsheet program like Excel.
For clarity, a summary of a simple CLI workflow is shown in the figure below.

The QUELO Command Line Interface (CLI): Installation¶
Installation Overview¶
Before you can run the QSimulate QUELO CLI, you need to install some infrastructure. This chapter describes the installation process. This is a straightforward process, only needs to be done once, and does not generally require administrative privileges.
The CLI will run from your local Linux host. Any standard modern Linux host can be used, including both dedicated Linux machines, as well as the “Linux Subsystem” that is supported in modern versions of Windows (versions 10 and 11). The CLI can also be installed and run from the Unix shell available as part of MacOS.
Installation requires that you download the “qysim” program package from QSimulate. You will download the package in whl (“wheel”) format, which can be installed using Python/Pip with a single command.
Before you install the qysim package, you will need to ensure you have the proper software infrastructure installed on your Linux installation. This is best carried out in a virtual environment, which serves multiple purposes:
This will allow you to install the necessary software without the need for “Administrator” privileges
This will ensure that the correct version of Python required to run the software is installed
This will isolate all the Python-installed software in a virtual container that cannot affect other software already installed on your computer
The implementation description that follows is for a Bash shell environment, which is the default shell for most Linux and Unix distributions. If you happen to use a different shell (e.g. tcsh, csh, etc.) you may need to modify the syntax of some of these commands, but the actual steps won’t change.
The process of installation is described in four parts:
Installation of miniconda3 (only performed one time; not necessary if already installed on your machine)
Creation of the QSimulate QUELO virtual environment
Installation of other software in the virtual environment that is required to run our software
Installation of the QSimulate software
Installing the virtual Environment (Miniconda)¶
If miniconda is already installed on your system, skip this section, and continue at “Creating the QSimulate QUELO virtual environment”.
We are going to set up the CLI access within the Miniconda environment management system. There are alternatives to Miniconda (e.g. Venv and PyDev), but Miniconda has advantages, particularly with respect to ensuring you don’t wind up with mixed (and conflicting) versions of Python on your system. Miniconda is an efficient reduced-size-and-scope version of the venerable Anaconda environment manager, and has all the features we need. You only need to install Miniconda once. If you have already installed Miniconda on your system, you won’t need to install it again now.
If you are an advanced user and prefer a different approach to the virtual environment, that’s fine–but you’ll need to modify the commands described below to reflect the approach you use, and you’ll be responsible for ensuring an appropriate version of Python (3.8 or higher) is installed and that it doesn’t conflict with other installed software. Unless you are an advanced user, we strongly recommend you use miniconda as described.
To install Miniconda3 run the following commands. This will create a subdirectory named miniconda3 in your home directory that will contain both the miniconda program, and, subsequently, any Python packages you install while in the virtual environment.
These installation instructions assume you are using the Bash shell.
1) Log into your Linux environment.
2) cd $HOME
3) mkdir -p ~/miniconda3
4) wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh \
-O ~/miniconda3/miniconda.sh
5) cd miniconda3
6) chmod ugo+rwx miniconda.sh
7) bash ~/miniconda3/miniconda.sh -b -u -p ~/miniconda3
8) rm -rf ~/miniconda3/miniconda.sh
9) ~/miniconda3/bin/conda init bash
10) cd $HOME
11) source .bashrc
Upon sourcing your .bashrc, you will find that your default Prompt: is replaced by “(base) Prompt:”, which reflects the fact that Miniconda is now installed and working on your machine. (base) Prompt: means that while Miniconda is running, you have not yet entered any named virtual environment. You can install software in the (base) environment, but it is generally considered bad practice and not recommended.
Creating the QSimulate QUELO virtual environment¶
Now that we have installed Miniconda, we can create virtual environments within Miniconda. Each virtual environment (VE) is an independent branch of your installed operating system. You can activate (enter) and deactivate (exit) a VE at any time. When you activate the VE, you can access all the Python packages you installed in that VE, but you won’t Python packages you may have installed in different VEs. As a result, Python-related software you install in a VE can’t pollute your system or break dependencies that are assumed for other packages installed on your computer outside the the VE. And if you make a mistake or don’t want to use a VE anymore, you can easily delete that VE–and all the Python packages installed to that VE–without affecting anything else on your computer.
For QUELO, we’ll create a VE named QUELO. In this VE, we’ll install a suitable version of Python and additional tools (Pip, Git, etc.) that are required for QUELO to install and run.
With these commands, we are creating a VE named QUELO. But you could use a different name, if desired. The conda create command creates the VE with a specific version of Python (3.8) that is suitable for our purposes. The conda activate command enters us into the created VE.
1) conda create --name QUELO python=3.8 -y
2) conda activate QUELO
After activating the QUELO environment your prompt should look like this:
(QUELO) Prompt_String:
where Prompt_String can vary, but is typically some combination of your login name, your machine name and your current path path, e.g.,
(QUELO) myname@mymachine:~$
Installing software in the QUELO virtual environment required to install/run QUELO¶
Before we install the actual QUELO package, we need to install some software that will be used during the installation process. The following commands take care of that installation. Be sure that you issued the “conda activate QUELO” command above, so that you are installing into the appropriate VE.
1) pip install -U setuptools setuptools_scm wheel
Installing QUELO (QYSIM)¶
1) Download the QUELO-specific QYSIM package using the link Qsimulate provides.
(This will be a .whl ["wheel"] Python installer package).
We will call this QYSIM.whl here,
but the actual name may vary and you should substitute that name below.)
2) (While in the QUELO VE) cd to the directory where you downloaded QYSIM.whl
3) pip install QYSIM.whl
4) hash -r
Using QUELO on subsequent logins¶
When you log out of your Linux session, all virtual environments are automatically closed. When you log in again, you will need to active (reopen) the VE you want to use. If you followed the installation instructions above, your VE for QUELO is, itself, named QUELO. To activate the VE to use it you need to issue the command:
conda activate QUELO
If everything is working properly, after issuing the above command, you should find your default prompt replaced as below
(base) Prompt:
(base) Prompt: conda activate QUELO
(QUELO) Prompt:
Testing the QUELO CLI¶
At this point, your CLI installation under the virtual environment should be working. To test it, try the following four commands (ensuring, first, that your QUELO virtual environment is active):
(QUELO) Prompt: sbb session set-url --url YOUR_QSIMULATE_ACCESS_URL
(QUELO) Prompt: sbb session login --user YOUR_QSIMULATE_EMAIL_LOGIN
(QUELO) Prompt: sbb batch list
(QUELO) Prompt: sbb session logout
Where YOUR_QSIMULATE_ACCESS_URL is replaced by the URL you were provided by QSimulate or your admin, and YOUR_QSIMULATE_EMAIL_LOGIN is replaced by the email address associated with your account. If your installation is properly set up, the first two commands above should log you into the platform (you should see “Login successful” after you specify your password following the login command). The batch list command should execute without an error. And the logout command will disconnect you from the session, and you should see “Logged out.” appear at your terminal.
Exiting the Virtual Environment¶
If you wish to exit the virtual environment, issue the command “conda deactivate”. This will drop you back to the (base) level of Miniconda.
(QUELO) Prompt: conda deactivate
(base) Prompt:
Removing the virtual environment and QUELO installation¶
One of the great advantages of having installed QUELO in a virtual environment is that it is straightforward to remove the installation. For example, if you wish to do a clean update installation, you can just remove the previous virtual environment, recreate the virtual environment, and then install the new version. (You can also have multiple virtual environments, if you wish to install the new update without removing the previous version).
If you have installed a number of different packages in the Miniconda environment manager, and only want to delete the QUELO installation (while keeping everything else as-is), you merely have to exit the VE using the command “deactivate” and then remove the VE, as follows:
(QUELO) Prompt: deactivate
(base) Prompt: conda env remove --name QUELO
(base) rm $HOME/.sbb_cli_rc
The last of these commands deletes a configuration file that QUELO will have created in your home directory.
To see a list of all virtual environments installed in Miniconda, you can use the “env list” command:
(base) Prompt: conda env list