QFP: Molecular Fingerprints¶
Overview¶
The molecular Fingerprints module allows the user to easily perform a complex workflow for a large number of molecules of interest. This workflow makes it very easy to generate 3-dimensional molecular structures from input SMILES strings, to perform conformational sampling for those 3D structures, to prune the resulting structures on the basis of energies and similarity, and to ultimately calculate quantum mechanical properties and 3D bitstring fingerprints for the resulting set of conformers. All of this is performed in an automated fashion that requires only the input SMILES data and a few button selections by the user.
A large number of quantum mechanical properties are determined for each molecule, providing a similar variety of properties to those presented in the well-known “QM9” and “QMUGS” database work.
The workflow that is performed is shown, at a high level, in the following figure.

The calculations that are performed are distributed across many processors on the cloud, enabling efficient performance, even for large numbers of input molecules. The platform can support a very large number of input SMILES strings in a single calculation. A major advantage of the QSimulate platform is that it has been developed to take advantage of the relatively less expensive spot instance pricing through cloud providers like AWS and Google Cloud in a manner that is seamless to the user.
This module is focused on high throughput analysis, and the expectation is that the user will download the resulting database at the end of the calculation. As a result, interactive tools to analyze the results in this panel are limited.
The Fingerprinting Task List¶
When you enter the platform, you will be presented with the Fingerprinting Task List, a list of calculations (Tasks) that you have previously set up and/or run, as well as a dialog to create a new Task. Clicking on a Task will bring you to the setup/results page for that Task.
For more details on the Fingerprinting Task List, see the chapter QSP Life User Interface.
Workflow Details¶
Overview¶
The workflow consists of two general steps: 3D conformer generation, and then energetic and similarity filtering. The filtering process can be seen as a funnel, where the top of the funnel has a larger pool of conformers that are progressively refined during each stage, to eventually lead to a smaller pool of distinct, low-energy structures. The funnel, which reduces the number of conformers being evaluated at each subsequent step, is designed to optimize throughput and reduce the number of conformers that are evaluated using the most computationally expensive approach to be applied (either semiempirical xTB or DFT quantum mechanics, depending on the option the user has chosen).
3D Conformer Generation¶
From the input SMILES string, a bounds matrix based on the input topology and atom types is calculated, and this is used as input to a distance geometry (DG) calculation. If, optionally, the user uploads a SDF file with structural information, then the bounds matrix is obtained from that input structure.
The bounds matrix, along with a random number seed, is used to create a specific distance matrix consistent with the bounds. This specific distance matrix is then used by DG to produce a starting structure. A series of N random number seeds are used to create N different specific distance matrices, which lead to N different DG-generated starting structures. The N structure pool is pruned to remove structures that are very similar to one another. Similarity among structures is evaluated using a root-mean-squared (RMS) coordinate metric.
Molecular Mechanics (MM) Optimization and Filtering¶
Starting with the conformer set from the first step (3D Conformer Generation) conformers are then subjected to a geometry optimization using the Universal Force Field (UFF).
Conformers with a MM energy larger than a cutoff value from the minimum identified are filtered out in this stage, as are structures that are too similar to another (lower energy) structure. The values used to filter the energy cutoff and the RMS similarity are automatically assigned by the program.
Semi-Empirical Quantum Mechanical (QM) Optimization and Filtering¶
The surviving conformers from the MM stage are then subjected to geometry optimization using the state-of-the-art semi-empirical QM method GFN-xTB. As with the MM step, high-energy and similar conformers are discarded from the pool, and the values that control these filters are defined by the program. Depending on the option chosen by the user, the set of conformers that survive the semi-empirical filtering are either used directly for QM property fingerprinting, or else are subjected to further filtering at the Density Function Theory (DFT) level.
Density Functional Theory (DFT) Optimization and Filtering¶
If the user has selected an option where QM properties will be evaluated at the DFT level, then the conformers that survive the semi-empirical filtering are optimized at the DFT QM level. As with the MM and semi-empirical steps, filtering is applied to retain only low energy distinct conformers. The DFT approach that is applied is, by default, (wB97x-D; def2-SVP). This is a level of DFT theory that optimizes throughput and reliability of the results.
QM Property Calculation (Characterization and Fingerprinting)¶
The set of conformers, as filtered by the above process, are ultimately sent for the final calculation of QM properties, termed “characterization.” A 3D bitstring fingerprint for each conformer is also determined, using the e3fp method. The complete list of properties that are calculated for each conformation is provided in the following table.
Note that output is provided in two formats. Most of the calculated properties are provided in a CSV format table. Coordinates are provided in SDF files.
Properties Calculated when each option set is chosen
==========================================================================================
Basic | Standard | Expert
==========================================================================================
Total Energy (xTB) | Total Energy (xTB) | Total Energy (DFT)
Atomic Energy (xTB) | Atomic Energy (xTB) | Atomic Energy (DFT)
Formation Energy (xTB) | Formation Energy (xTB) | Formation Energy (DFT)
Dipole Moment (xTB) | Dipole Moment (xTB) | Dipole Moment (DFT)
Quadrupole Moment (xTB) | Quadrupole Moment (xTB) | Homo Energy (DFT)
Homo Energy (xTB) | Homo Energy (xTB) | Lumo Energy (DFT)
Lumo Energy (xTB) | Lumo Energy (xTB) | Homo-Lumo Gap Energy (DFT)
Homo-Lumo Gap Energy (xTB)| Homo-Lumo Gap Energy (xTB) | IAO Charges (DFT)
Fermi Level (xTB) | Fermi Level (xTB) | XC Energy (DFT)
Mulliken Charges (xTB) | Mulliken Charges (xTB) | Nuclear Repulsion Energy (DFT)
Solvation energies (xTB) | Solvation energies (xTB) | One Electron Energy (DFT)
| Total Free Energy (xTB) | Two Electron Energy (DFT)
e3fP fingerprint (xTB) | Enthalpy (xTB) |
3D Coordinates (xTB) | Heat Capacity (xTB) | +All props in "Standard" (xTB)
| Entropy (xTB) |
| | e3fp fingerprint (DFT)
| e3fp fingerprint (xTB) | 3D Coordinates (DFT)
| 3D Coordinates (xTB) |
| |
| |
| |
------------------------------------------------------------------------------------------
Fingerprinting Options¶
In this section, you will select the options that control what descriptors are calculated, as well as the computational approach. In addition, if you have selected “Expert” mode in your user-options panel, you can also designate if multiple conformers will be generated for each input molecule, and the filtering options that will be applied to the generated conformers.
Broadly, in terms of the computational method used, fingerprints and properties can be calculated using either the fast and inexpensive semiempirical approach, or using the more precise but slower and more computationally expensive DFT method. The selection of which approach to use will often depend on how many molecules you wish to characterize and how quickly you wish to get back the results. If you have a lot of molecules (many hundreds or more) and/or you need the results as quickly as possible, you may wish to use the semi-empirical method. If, on the other hand, you either aren’t characterizing a large number of molecules or you want the best possible predictions and can wait for those to finish, DFT is often the better choice. DFT calculations typically take several orders of magnitude more compute time and provide several orders of magnitude less throughput, and, as a result, if you wish to characterize a very large number of molecules, you will typically avoid DFT.

There are three options for fingerprinting/property calculation. You can select only one, and you choose it by clicking on the box corresponding to your choice. A full list of the properties calculated with each option is shown above in the section “QM Property Calculation (Fingerprinting)”. Here, we describe the calculation options in general terms.
GFN2-xTB (basic): All QM properties will be determined using the GFN2-xTB semi-empirical QM approach. Because DFT is not being used, this saves computational expense and increases computational through not only at the property calculation stage, but also because the final DFT-level filtering step can be skipped. A list of properties that are calculated is given in the Workflow Details section. In Basic mode, the more costly vibrational properties (free energy, etc) are skipped.
GFN2-xTB (full): All QM properties will be determined using the GFN2-xTB semi-empirical QM approach. In addition to the properties calculated in Basic, properties dependent on vibrational analysis will also be calculated.
DFT: QM properties will be determined using the DFT approach, using the default DFT approach, which is (wB97x-D; def2-SVP). This level of DFT theory generally provides very good results at a reasonable of cost. A list of properties that are calculated is given in the Workflow Details section. DFT filtering is applied in the conformer refinement workflow, after the xTB semi-empirical step (see the workflow above). In addition a large number of DFT properties, all the xTB properties listed for “Standard” will also be calculated and reported.
Fingerprinting Options: Expert Mode¶
If you have selected “Enable the Expert Mode” from your Account Management dialog, an additional dialog will appear in Options Panel. Below is the options panel in Expert Mode:

The additional options in this Expert view relate to the generation and filtering of conformers for each input molecule. The default behavior for the program is to expect 3D structure files in SDF format, which are passed as-is to the platform for calculation of 3D fingerprints and properties.
If you wish to generate conformers from the input molecules, then you have two choices for input file type, SMILES and SDF, and in either case the platform will generate multiple conformers for each molecule, and then filter/reduce the initial conformer set for each molecule according to cutoffs that you can specify here. Unless you are interested in using QFP specifically for rescoring, conformer generation is strongly recommended.
Generate Conformers:
Toggle box unchecked (default): Input molecules are used as supplied, and passed directly to the software nodes that will generate a 3D fingerprint and the chosen descriptors. Since no conformers are being generated, in this case only SDF format input with 3D coordinates is accepted.
Toggle box checked: For each input molecule, a set of conformers will be generated using distance geometry (DG). Either SDF or SMILES input is acceptable in this case. If this toggle is checked, additional options will be available, as described below. These options are not available if the toggle is not checked.
Max conformers generated: The maximum number of conformers to be generated for each input molecule. The default is 50 if the Generate Conformers toggle box is checked. Conformers are randomly generated using DG using a bounds matrix obtained from the input molecule. A larger value here results in a more thorough exploration of conformational space, but at a higher calculation cost per molecule.
Min conformers generated: The minimum number of conformers to be generated for each input molecule. The default is 1 if the Generate Conformers toggle box is checked. The total number of generated conformers will depend on the size of each molecule, but it will be typically determined by the maximum given (described above).
Max conformers retained: The maximum number of conformers that is retained after RMSD and energy filtering (described below). If the remaining number of conformers is lower than this maximum, all of them are retained. Otherwise, a hierarchical clustering will be performed to remove conformers until the user-defined maximum number is reached.
RMSD Threshold (Angstroms): The RMSD threshold used to filter out conformers that are too similar to each other. If the RMSD between the heavy atom coordinates of two conformers of a molecule is lower than the RMSD Threshold, the higher energy structure is removed from the set.
Energy Threshold (kcal/mol): The energy threshold used to filter out conformers with too high an energy. If the difference between the energy of a conformer and that of the lowest-energy conformer found is higher than this threshold, the conformer is removed from the set.
Compound Input Requirements¶
QFP expects compounds to be provided with an interpretable Lewis structure. Before uploading the input files for your compounds, please check that they all conform to the following:
Is a single stereoisomer with all chiral centers defined
Describe singlet ground states
If using SDF
All atoms are present, fully described, and consistent with charge state (including hydrogen)
If using SMILES
Formal charges are assigned to atoms bearing them
Further descriptions of these requirements can be found below.
Treatment of Undefined Stereoisomers¶
Stereoisomer ambiguities are not allowed. If using SMILES, the chirality of stereocenters in a molecule must be specified in the input SMILES string.
For example, the following input is not accepted:
CCC1C[N+]CCC1F
The following, with fully specified stereochemistry, is:
CC[C@@H]1C[N+]CC[C@@H]1F
A SDF format file, if supplied, must contain a full coordinate definition of the input molecule.
If you are unsure about the stereochemistry of your compound, we recommend generating any/all stereoisomers and submitting each as a separate compound.
Excited States and Unusual Charge States¶
QFP currently expects every compound to be in a singlet, ground state. It does not support property generation for:
Radicals
Compounds with broken valence (carbon forming 5 bonds, etc.)
Specification of Formal Charge and Unusual Charge States¶
When using SDF files, the file must fully conform to the V2000 or V3000 format. All formal charges must be assigned to atoms to describe the total formal charge state of the compound.
When using SMILES, formal charges must be defined by placing them on the appropriate atoms. For example:
C([N](=O)[O]) (incorrect)
C([N+](=O)[O-]) (correct)
Both Implicit and explicit hydrogen are allowed, but note that explicit hydrogen are not a sufficient replacement for a full description of the formal charge. Charge state is defined based on the proscribed formal charges not on hydrogen.
For example, the SMILES below describes a carboxylate:
CC(=O)[O-]
While this SMILES instead describes the protonated carboxcylic acid with an implicit hydrogen:
CC(=O)O
In cases where the explicit hydrogens are inconsistent with the formal charge the platform will reject the input. The following example would be rejected as the charge state of the amine is ill-defined - it is unclear whether the amine should have one less hydrogen or should be charged.
CC[C@@H]1C[N]([H])([H])CC[C@@H]1F
Any of the following are acceptable, as they fully describe the protonation and charge state of that amine.
CC[C@@H]1C[N+]CC[C@@H]1F
CC[C@@H]1C[N+]([H])([H])CC[C@@H]1F
CC[C@@H]1C[N]CC[C@@H]1F
CC[C@@H]1C[N]([H])CC[C@@H]1F
Please note that unusual charge states often struggle to find a converged structure and generate properties. An example of this is a carbon bearing a negative formal charge. If you encounter issues with a compound repeatedly failing to generate properties, first confirm that its charge state is correct and that there are not other ways to describe its structure that better conform to a standard Lewis structure (like removing separation of charge). After confirming this, generating more conformers by setting Max conformers generated to a much larger value may help.
Structure Input¶
Molecules are imported in the platform in the “File Uploads:” section.
Allowed Input File Formats¶
The file formats allowed in this section will depend on the whether you have modified the default Conformer Generation toggle option in the previous section. By default, conformers are not generated, and in this case, only SDF 3D structure input is allowed (and the SMILES radio button option will not appear). If you have clicked the Conformer Generation toggle, then you have a choice of either SMILES or SDF input (chosen via radio buttons above the file specification box). SMILES input is provided as one molecule per line. SDF input files can contain multiple concatenated molecules in a single file.
Uploading the Molecules File¶
Clicking on “Browse” opens the file browser on the host computer. Once a file is selected, you click on the Upload button to parse the file. If the number of uploaded molecules is <= 50, then an interactive table will be presented, as shown below. If more than 50 molecules are input, then the table is not shown, to reduce memory overhead on the browser. In the latter case, you can still download the import report using the Download Report button.
User view if number of SMILES <= 50:

User view if number of SMILES > 50:

You can upload molecules from multiple files, if desired, by executing the browse/upload process repeatedly.
Supported SMILES Format¶
SMILES format is one SMILES string per line, with a user-supplied name for the SMILES string optionally provided:
The SMILES string must not contain space characters. A string of one or more space characters separates the SMILES string from the (optional) NAME. NAME is an alphanumeric string that will be used in the status and output parts of the panel. NAME is optional, and if not supplied, a name will automatically be assigned by the platform, using the format LNNN, where NNN is a numerical index that is applied for all input ligands without names, starting from L001. This index is extended as needed to accommodate a larger number of compounds uploaded without names (i.e. L1000, L10000).
For example, the input for a list of 5 amino acids would be:
N[C@H](C(=O)O)C ALA
N[C@H](C(=O)O)CCCNC(=[NH2+])N ARG
N[C@H](C(=O)O)Cc1ccc(cc1)O TYR
N[C@H](C(=O)O)CCC(=O)N GLN
N1[C@H](C(=O)O)CCC1 PRO
which corresponds to the amino acids: ALA, ARG, TYR, GLN, PRO.
If you specified the SMILES without the optional names after the SMILES strings, then these five molecules would be internally assigned the names L001, L002, L003, L004, and L005.
Supported SDF Format¶
SDF files can include a concatenation of multiple SDF definitions. Standard SDF format files are expected. If the SDF input is being used without conformer generation, then it is required that the SDF file contains 3D coordinates for each molecule.
Parsing/Validation Check (<= 50 Molecules)¶
The specified file with the SMILES definitions will be checked for validity once you click on the “Upload” button. If the number of input SMILES is <= 50 (interactive mode), you will also have the possibility, if desired, to remove any SMILES that was successfully imported by clicking on the red cross next to the SMILES name. Clicking on any row of the table corresponding to a successfully imported structure will present the 2D representation of the structure to the right of the table:

For the example here, an invalid SMILES was intentionally included in the input file to demonstrate the program behavior when that is identified. The compound (named “BadCmpd” in the input file) appears as a red shaded line. If you click on that compound, information on the error will appear in the Information field.

Note: The SMILES processing assumes closed shell calculations, therefore the multiplicity is always set to 1.
Parsing/Validation Check (> 50 Molecules)¶
In the case of an upload of more than 50 molecules, instead of an interactive table, only a summary of the molecules uploaded will appear. This table indicates the numbers of Valid and Invalid molecules uploaded, plus the Total of the two values. You can use the Download Report button (below) to examine why any molecules were deemed Invalid. You can also use buttons below the table to either delete the invalid uploaded molecules, or else to delete the entire set of uploaded molecules.
Download Report¶
The Download Report button will download the information in the structures table, in .csv format. The columns in the table are NAME, SMILES, STATUS, and MESSAGE. MESSAGE is blank unless the STATUS indicates a problem processing the SMILES string.
Delete Invalid¶
This button only appears when the number of molecules imported is > 50. In this case, the Delete Invalid button can be used to delete any molecules flagged as Invalid from the set. Note that if you don’t delete this molecules from the set, you can still submit/run the calculation–the molecules flagged as “Invalid” will simply be skipped.
Delete Uploaded¶
This button only appears when the number of molecules imported is > 50. In this case, the Delete Uploaded button can be used to delete all uploaded molecules.
Starting the Calculation¶
Once you have uploaded your data, chosen the fingerprinting level, and specified any other options of interest, you can start the calculation by pressing the Start Simulation button, which appears below the Fingerprinting Options selector.
Simulation Status¶
After the calculation has been started, you can monitor the status in the section of the panel that appears below the Options section. In addition to information about how much computer time has been used (vCPU usage), you can also Stop and Resume a calculation that is in progress, if necessary. Stop will terminate the calculation but keep the intermediate files so that you can subsequently resume the calculation, if desired.

The Simulation Status will update regularly at 3 minute intervals. If you wish to update more frequently, you can click on the indicated button beneath the Manual Update section.
Beneath the progress bar on the right, you will find a summary list of how many compounds are in each part of the calculation workflow. This provides a more detailed view of the calculation progress.
Diagnostic Report¶
If any compounds fail to generate properties for any reason during the calculation, a diagnostic report of additional information will appear under the Summary section of Overall Status.
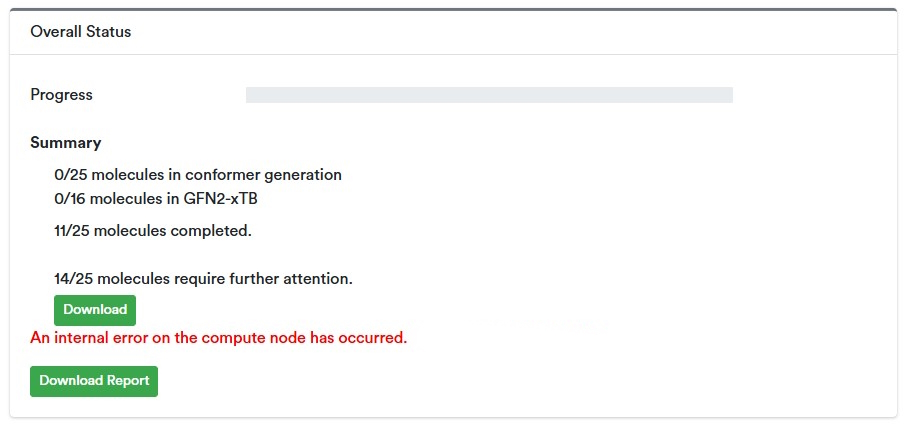
This includes the number of compounds that need further attention, a “Download” button that yields a csv of each compound with a message explaining why it failed to generate properties, a red message relevant to the calculation (rather than any specific compound), and finally the “Download Report” button that retrieves a zip file of jsons containing more details on the runtime options. All this information can assist in troubleshooting and should be shared with technical support as necessary.
Results¶
Once the calculation has completed, the buttons in the Results section will become active. In this section you can download the results of the calculation. You can also examine the results for any particular ligand by using the search bar.

There are a few options for this download:
Max. number of conformers: The maximum number of conformers to be reported for any input molecule. Because of the filtering process performed in the workflow, the number of conformers for each molecule will vary, and may be smaller than the number specified. If the number of conformers post-filtering is larger than the maximum value specified (NMAX), the NMAX lowest energy conformers will be reported. If NMAX=1, then only the single lowest energy conformer for each molecule will be reported.
Energy Units: The units to be used for any predicted energetic property. The default is Hartrees, and kcal/mol is also available.
Show all properties: If checked, the downloaded csv will include columns for all possible predicted properties. This includes properties that were not predicted if a smaller property set was selected. The unpredicted properties will appear as blank columns with just the header label.
Include incomplete molecules: If checked, molecules that failed to return predicted properties will also appear in the downloaded csv. If unchecked these are omitted from the results download csv entirely.
Results Downloaded ZIP file¶
The Download button is only active when the Calculation has the status of “Complete”. When you press the “Download” button, a system-dependent dialog will ask where to a “.zip” formatted file. This .zip file contains the CSV formatted spreadsheet of calculated properties (see below), as well a directory tree of SDF structure files corresponding to all the conformers reported in the table. The format of the directory tree is
results.csv
structures [directory]
[SMILES name 1]
xtb_results_0.sdf
...
xtb_results_M.sdf
dft_results_0.sdf
...
dft_results_N.sdf
input.smi
[SMILES name 2]
xtb_results_0.sdf
...
xtb_results_M.sdf
dft_results_0.sdf
...
dft_results_N.sdf
input.smi
[etc.]
A sub-directory for each input molecule is included in the .zip file. The list of xtb_results structures are the M conformers that survived the workflow filtering (up to a maximum of NMAX, as specified by the user). The dft_results files are included only if DFT calculations were performed (Expert), and in this case, the N conformers that survived the workflow filtering (up to a maximum of NMAX, as specified by the user) are provided. input.smi contains the input SMILES string for this molecule.
Results Spreadsheet¶
The results.csv file included in the downloaded .zip file contains the calculated QM values for each molecular conformer. This standard-format “.csv” file can be read by Microsoft Excel, Google Sheets, or LibreOffice (or any other programs that handle this format).
A portion of the spreadsheet, viewed in Microsoft Excel, is shown below. There is one line (entry) for every conformer of every input molecule, so there will often be multiple lines with the same SMILES and name (SMILES_tag). The second column is “ID”, which gives the conformer number for the parent SMILES, starting from 0. Note that the number of conformers may differ for each SMILES, but will not exceed the value of NMAX specified when requesting the download. If NMAX is specified as “1” (only download the lowest energy conformer), then the ID of each molecule would be “0”.

Results Search¶
If you enter the name of an input molecule into the search bar and click on the search button, details for that molecule will populate the bottom of the panel (if you enter a name that does not correspond to any output, then the Results Preview area will remain blank). Below the name of the molecule, you will find a series of dark grey boxes. Each of these corresponds to one of the conformers generated for that molecule that passed all the filtering tests in the workflow. (Conformers that were removed at some filtering stage are not included here). The conformers are listed in order of descending energy, with the lower energy to the right. The conformer with the lowest energy is always the last in the list, and has a thin red border around it. Ordering is based on DFT energy (if DFT calculations were performed), or else the GFN-xTB energy.
One box will have a thick green border around it. This is the selected conformer. You can click on any box to change the selection. The table and 3D view of the conformer below reflect the chosen conformer. The scrollable table will contain the calculated results for the chosen conformer. (The same information appears in the downloaded .csv file).
The molecule view in the 3D viewer can be adjusted using either left-click or right-click to rotate and the scroll wheel to resize.
A sample conformer view is shown below:

Below the 3D viewer, buttons appear to allow you to either Download the coordinates of the shown molecule (in .xyz format) or else to copy them to the clipboard associated with your browser. You will also find a Reset button that resets the view in the visualizer.
Molecular Fingerprint Property Definitions¶
Here you will find a short description of every property calculated by this module. This expands upon the table in the main Fingerprinting chapter, which lists these properties by name, but does not provide descriptions.
============================================================================================
Property B S E Description
============================================================================================
NAME |X|X|X|Original Compound Name
ID |X|X|X|Conformer ID
SMILES |X|X|X|Original SMILES
GFN2_ENERGY |X|X|X|Total GFN2 electronic energy
GFN2_DIPOLE_X |X|X|X|x-component of the electronic dipole
GFN2_DIPOLE_Y |X|X|X|y-component of the electronic dipole
GFN2_DIPOLE_Z |X|X|X|z-component of the electronic dipole
GFN2_DIPOLE_TOT |X|X|X|Total magnitude of the electronic dipole
GFN2_HOMO_ENERGY |X|X|X|Energy of the highest occupied molecular orbital
GFN2_LUMO_ENERGY |X|X|X|Energy of the lowest unoccupied molecular orbital
GFN2_HOMO_LUMO_GAP |X|X|X|Energy difference between HOMO and LUMO
GFN2_QUADRUPOLE_XX |X|X|X|x,x component of the electronic quadrupole
GFN2_QUADRUPOLE_XY |X|X|X|x,y component of the electronic quadrupole
GFN2_QUADRUPOLE_YY |X|X|X|y,y component of the electronic quadrupole
GFN2_QUADRUPOLE_XZ |X|X|X|x,z component of the electronic quadrupole
GFN2_QUADRUPOLE_YZ |X|X|X|y,z component of the electronic quadrupole
GFN2_QUADRUPOLE_ZZ |X|X|X|z,z components of the electronic quadrupole
GFN2_FERMI_ENERGY |X|X|X|Fermi energy level
GFN2_SOLVATION_ENERGY_ACETONITRILE|X|X|X|Energy difference between GFN2 structure in acetonitrile and vacuum using PCM model
GFN2_SOLVATION_ENERGY_THF |X|X|X|Energy difference between GFN2 structure in THF and vacuum using PCM model
GFN2_SOLVATION_ENERGY_WATER |X|X|X|Energy difference between GFN2 structure in water and vacuum using PCM model
GFN2_E3FP |X|X|X|Molecular Fingerprint from the GFN2-minimized structure via the E3FP Method
GFN2_ENTHALPY_ROT | |X|X|Enthalpy from rotational degrees of freedom
GFN2_ENTHALPY_TRANSL | |X|X|Enthalpy from translational degrees of freedom
GFN2_ENTHALPY_VIB | |X|X|Enthalpy from vibrational degrees of freedom
GFN2_ENTHALPY_TOT | |X|X|Total enthalpy
GFN2_ENTROPY_ROT | |X|X|Entropy from rotational degrees of freedom
GFN2_ENTROPY_TRANSL | |X|X|Entropy from translational degrees of freedom
GFN2_ENTROPY_VIB | |X|X|Entropy from vibrational degrees of freedom
GFN2_ENTROPY_TOT | |X|X|Total entropy
GFN2_HEAT_CAPACITY_ROT | |X|X|Heat capacity from rotational degrees of freedom
GFN2_HEAT_CAPACITY_TRANSL | |X|X|Heat capacity from translational degrees of freedom
GFN2_HEAT_CAPACITY_VIB | |X|X|Heat capacity from vibrational degrees of freedom
GFN2_HEAT_CAPACITY_TOT | |X|X|Total heat capacity
DFT_ENERGY | | |X|Total DFT electronic energy
DFT_XC_ENERGY | | |X Exchange-correlation component of DFT electronic energy
DFT_NUCLEAR_REPULSION_ENERGY | | |X|Nuclear repulsion component of the DFT electronic energy
DFT_ONE_ELECTRON_ENERGY | | |X|One-electron component of the DFT electronic energy
DFT_TWO_ELECTRON_ENERGY | | |X|Two-electron component of the DFT electronic energy
DFT_DIPOLE_X | | |X|x-component of the electronic dipole
DFT_DIPOLE_Y | | |X|y-component of the electronic dipole
DFT_DIPOLE_Z | | |X|z-component of the electronic dipole
DFT_DIPOLE_TOT | | |X|Total magnitude of the electronic dipole
DFT_HOMO_ENERGY | | |X|Energy of the highest occupied molecular orbital
DFT_LUMO_ENERGY | | |X|Energy of the lowest unoccupied molecular orbital
DFT_HOMO_LUMO_GAP | | |X|Energy difference between HOMO and LUMO
DFT_SOLVATION_ENERGY_ACETONITRILE | | |X|Energy difference between DFT structure in acetonitrile and vacuum using PCM model
DFT_SOLVATION_ENERGY_THF | | |X|Energy difference between DFT structure in THF and vacuum using PCM model
DFT_SOLVATION_ENERGY_WATER | | |X|Energy difference between DFT structure in water and vacuum using PCM model
DFT_E3FP | | |X|Molecular Fingerprint from the DFT-minimized structure via the E3FP Method
============================================================================================
* B = Basic
* S = Standard
* E = Expert
The Molecular Fingerprints Command Line Interface (CLI)¶
The CLI vs the GUI¶
The Molecular Fingerprints Command Line Interface (CLI) is an alternative way to access the functionality of this platform. It provides the same functionality as the GUI version that is described in previously chapters, and it is not required that you either install or use the CLI. Whether you use GUI or CLI access is entirely a matter of preference. Note that the CLI requires you install local software in a Python environment, and so it is limited to platforms where you have ready Linux access, and where you can install some requisite software.
In contrast, the GUI is accessed through a standard browser and requires no software installation. Therefore, GUI access is available on a much larger array of devices (computers, tablets, phones, etc.).
Note that both CLI and GUI initiated calculations are stored on the backend in the same databases. That means you can access calculations that were initially run from the CLI through the GUI, and vice-versa. This also means your account and credentials are the same between the CLI and GUI.
Installation Note¶
Note that before you can run the CLI, you need to have installed the CLI on your computer, following the instructions provided in the chapter The Molecular Fingerprints Command Line Interface (CLI): Installation. Installation only needs to be performed once (unless you need to update the software). If you want to use the CLI, you must install the software first, using the step-by-step instructions in that chapter.
Assuming you have followed the instructions in the installation chapter and installed the QuantumFP CLI into a virtual environment under Miniconda, each time you log into the computer and want to access the CLI, you will need to activate the appropriate virtual environment that you created.
Overview¶
The previous chapters have described the functionality of the Molecular Fingerprints platform, and how to access the platform through the browser-based GUI. As noted, an identical set of functionality is accessible through the CLI.
The CLI runs in the Linux environment. To run the CLI, some infrastructure must be installed using a small number of simple commands, as are described in the Installation section (next chapter). Once the CLI is installed, the user can invoke it using the sbb command. The available sbb commands and syntax are described in the sections below.
For details on implementation of the QuantumFP process, the user is referred to previous chapters.
In the sections that follow, the following should be noted:
Enclosing arguments with double quotes is required for arguments with space or other special characters, e.g. a batch name with embedded spaces.
Optional flags are enclosed in square braces.
Flags that require arguments are followed by a value in angle braces.
Running the QSimulate CLI¶
Once you have installed the CLI package, following the instructions in the Installation chapter, and you have activated the appropriate virtual environment you will be able to run calculations from the command line. The software is invoked using the “sbb” command, followed by options and keywords that describe exactly what you want to do. For example, sbb -h will return a top-level help menu:
(QuantumFP) Prompt:~/Desktop$ sbb -h
usage: sbb [-h] [-V] [--rc RC] Commands ...
CLI interface for the QSimulate Platform
positional arguments:
Commands
session Logging in, logging out, setting the server address
batch Create a batch job and set options for the job, run or delete a batch
molecule Attach molecule file(s) to a created batch
options:
-h/--help Show this help message and exit
-V/--version Show version and exit
--rc RC Set an alternate config file
Session Command¶
Session Set-URL¶
Logging into the CLI requires that you fist specify the name of the server you will be using for your calculations, and then actually logging into that server.
To specify the URL of the server (a URL you will have been provided by QSimulate or your system administrator) use the following command. Note that this URL will be just the domain name without http or https. This command only needs to be performed once during a session, even if you log out and back in again during that session.
sbb session set-url URL_TO_ACCESS_QSIMULATE
[For example: sbb session set-url my-company.qsimulate.com]
Session Login and Logout¶
To log into the server, you use the command
sbb session login --user USERNAME -p PASSWORD
USERNAME and PASSWORD are replaced by the credentials you set up for the QSimulate platform. Note that these are exactly the same as the ones you use to access your account through the GUI. If you do not wish to write your password in plaintext in the command line, you can omit the “-p PASSWORD” part of the command, in which case the server will prompt you to enter your password.
If your login was successful, you will see the message
Login successful.
To subsequently logout of the server, use the command
sbb session logout
Note that you will be auto-logged off the server after 15 minutes of inactivity. If you attempt to issue a sbb command that requires a response from the server after you have been logged out, you’ll be shown the message
Please login
Session Update-Certificate¶
It is unlikely you’ll ever need to manually update the SSL certificate. But if you do, the certificate can be updated using the command
sbb session update-certificate
Batch Command¶
Within the QSimulate platform, calculations you want to run are termed “batches”. Each calculation run is a “batch”. The batch command can be used to list and/or examine batch calculations you have already submitted, or to create and submit a new batch calculation. The main level options for batch command are:
sbb batch [create]
[run]
[create-and-run]
[list]
[results]
[delete]
[stop]
[restart]
[options]
The standard workflow for setting up a calculation requires three commands:
sbb batch create
sbb molecule create
sbb batch run
The first sets the options for the calculation. The second attaches molecular input files. The third submits the calculation for execution. There is also a sbb batch create-and-run command that combines all actions into one command.
Important Considerations for Scripted Execution¶
If you plan to run the CLI commands from a script or pipe, it is crucial to understand that while the molecule create command will return the user immediately to the command line, the actual upload and attachment of the molecules file can take some time. During this time, the status of the job will be “busy”. If you attempt to run a job while it is in “busy” status, you will get an error message indicating you must wait for the job to proceed to “staged” status. While you can just wait for the molecule upload to complete and reissue the run command, this is often inconvenient in a scripted implementation. To circumvent this issue, the user has two options:
Use the batch-create-and-run command (see below) in place of the separate batch create/molecule create/batch run commands. This is the recommended alternative, and retains all the options available to the user with the three separate commands.
Insert script code in your submission workflow between molecule create and batch run to poll the batch status (via a batch list command), to ensure that it has exited the “bush” status and is instead in the “staged” status.
The various commands are detailed in the sections below
Batch Create¶
sbb batch create
-b/--batch <batch_name> \
[-t/--type <basic/standard/expert>] \
[--no-generate_conformers/--generate_conformers] \
[--max_conformers <num_conformers> ] \
[--rthresh <rthresh> ] \
[--ethresh <ethresh> ] \
[--dry-run ]
Calculation options for a batch are configured when this command is called. A set of calculation options is specified with the -t switch, and additional advanced options can be specified with further command-line switches. To fully set up a calculation, a “batch create” command needs to be followed by a “molecule create” command (below), and a subsequent “batch run” command will run the calculation.
sbb batch create options detail:
-b/--batch <batch_name> \
Required. Defines the name of a new batch name to be created.
-t/--type <basic/standard/expert>
Specifies the set of descriptors and fingerprints to be generated. There are three options, corresponding to the three option buttons in the GUI version: Basic, Standard, and Expert. (Additional details of what, specifically, is included in each set is described in separate chapters).
-t basic: Basic fingerprinting options, GFN2-xTB optimization, GFN2-xTB 3D fingerprint, GFN2-xTB electronic properties
-t standard: Standard fingerprinting options: GFN2-xTB optimization, GFN2-xTB 3D fingerprint, GFN2-xTB electronic properties, GFN2-xTB vibrational properties.
-t expert: Standard fingerprinting options: GFN2-xTB optimization, GFN2-xTB 3D fingerprint, GFN2-xTB electronic properties, GFN2-xTB vibrational properties, DFT optimization, DFT 3D fingerprint, DFT electronic properties.
The remaining properties are termed “Expert” properties in the GUI interface, and are only shown in “Expert Mode”. They can be set here using the following options.
--no_generate_conformers (DEFAULT)
--generate_conformers
Controls whether conformations will be generated. The default is “no_generate_conformers”. This default expects the input molecule data in SDF format, with the 3D conformation of each molecule pre-determined. No conformational exploration will be performed, and descriptors and fingerprints will only be calculated for the input conformations. This option is not compatible with 2D SMILES input.
If you want conformers to be generated for each input molecule (optional for sdf format input, and required for SMILES input format) you need to specify the generate_conformers flag. A total of NUM_CONFORMERS are initially generated for each input molecule.
--max_conformers <num_conformers>
Default is num_conformers = 50. Specifies the maximum number of initial conformations to be generated for each molecule in the input list. Ignored if no_generate_conformers has been specified.
--rthresh <rthresh>
Default is rthresh = 0.1 (Angstrom). Defines the RMSD threshold for discarding conformationally redundant conformers during the filtering process. For conformer pairs where the RMSD is less than rthresh, the higher energy conformer will be discarded. Ignored if no_generate_conformers has been specified.
--ethresh <ethresh>
Default is ethresh = 10.0 (kcal/mol). Defines the energy threshold for discarding high energy conformers during the filtering process. Any conformer that is more than ethresh higher in energy than the lowest energy conformer identified will be discarded. Ignored if no_generate_conformers has been specified.
--dry-run
If the dry-run argument is specified, the calculation options will be written to standard out in JSON format and no batch calculation will be created.
Batch Run¶
sbb batch run -b/--batch <batch_name> [--priority <priority>]
This command will submit (run) a batch job that was previous created and has the status of “staged”. Note that before you can run a job, you must associate a molecules input file to that job using the Molecule Create command (see below). If you attempt to run a batch for which you have not defined input molecules, you will get an error message indicating “no valid inputs are present.”
If you attempt to issue the batch run command before the molecule upload is complete (i.e. a molecule create command that is not finished executing), you will receive an error message indicating the batch status is “busy” and must be “staged” before it can be run. In this case, you must wait for the molecule upload to finish before issuing the batch run command. For unattended script-based submission, it is strongly recommended that instead of issuing the batch create/molecule create/batch run commands separately, you use the batch create-and-run command, which circumvents this type of issue.
--priority <priority>
Default is 75. The queueing priority of the jobs, relative to other jobs the same user/company has submitted. The priority can be used for ensuring a particular job gets sent for execution before others in the user’s queue. It has no affect at all on performance/turnaround once the job exits the queue and starts executing. The default priority for CLI jobs is 75, while the default CLI for GUI-submitted jobs is 50. Lower values mean higher priority. If you want CLI jobs to execute before GUI-submitted jobs, assign priorities < 50. The minimum allowable value is 10.
Batch Create-and-run¶
sbb batch create-and-run \
-b/--batch <batch_name> \
-f/--file <molecule_filename_and_path> \
[-m/--molecule-type <molecule-type>] \
[-t/--type <basic/standard/expert> ] \
[--no-generate_conformers/--generate_conformers] \
[--max_conformers <num_conformers> ] \
[--rthresh <rthresh> ] \
[--ethresh <ethresh> ] \
[--priority <priority>]
This command is entirely analogous to a combination of “batch create”, “molecule create”, and “batch run”, but in this case, the batch job is both set up and run in a single command. It eliminates the need to issue multiple commands. The individual options are as described under Batch Create and Molecule Create.
It is strongly recommended that you use the batch create-and-run command if you will be running jobs from a script, since it will automatically wait for the molecule upload to finish before issuing the run command.
Batch List¶
sbb batch list [-o/--output-style <table/csv/html/json>]
Returns a list of all batch calculations that have been created and/or run. All batch calculations run from the account will be shown, including calculations (if any) performed using the GUI interface.
The optional output-style specifier will designate the format of the list. The default is “table”. An example output table would be:
+---------------+--------+----------+
| Batch Name | smiles | status |
+---------------+--------+----------+
| Test Batch 01 | 2 | complete |
| Test Batch 02 | 2 | staged |
+---------------+--------+----------+
The same output in csv format (sbb batch list -o csv) would be:
Batch Name,smiles,status
Test Batch 01,2,complete
Test Batch 02,2,staged
html format is intended for use in Jupyter notebooks.
Potential status values include: staged, running, complete, stopped, failed, and busy.
Staged: Job has been created but not submitted to run. Or a previously stopped job in the process of resuming.
Running: Job is currently in process on the servers
Complete: Job has completed successfully
Stopped: Job was paused using the batch stop command
Failed: Job failed to complete successfully
Busy: A staged job for which a molecule upload is in progress
Batch Results¶
sbb batch results -b/--batch <batch_name> [-o/--output-style <table/csv/html/json>]
This command will return the results for the completed batch calculation with the name batch_name. If batch_name includes embedded spaces, you must enclose the full name with double quotes. The result is streamed to standard out, and you will typically redirect to a file. For example:
sbb batch results -b "Test Batch 01" --output-style csv > batch01.csv
The -o/-output-style format operates exactly as described above for Batch List.
Batch Delete¶
sbb batch delete -b/--batch <batch_name>
This command will delete the named batch. Note that the delete will be performed immediately and there is no subsequent user verification, so be careful when using this command.
Batch Stop¶
sbb batch stop -b/--batch <batch_name>
This command will stop the named batch. The state of the calculation and all intermediate results are retained when the calculation is stopped, and it can subsequently be restarted using the batch restart command (below). This command can only be successfully be executed for a job whose current status is “Running”.
Batch Restart¶
sbb batch restart -b/--batch <batch_name>
This command will restart a previously stopped batch. This command can only be successfully be executed for a job whose current status is “Stopped”.
Batch Options¶
sbb batch options -b/--batch <batch_name>
This command will report the options used for the named batch in JSON format. The report is sent to standard output.
For example, a batch job run with all default options would return this JSON report:
{
"jobType": "qsp_fingerprinting",
"type": "standard",
"options": {
"generate_conformers": false,
"max_conformers": 50,
"rthresh": 0.1,
"ethresh": 10.0
}
}
Molecule Command¶
Molecule Create¶
sbb molecule create -b/--batch <batch_name> \
-f/--file <path-to-file> \
[-t/--type <type>]
This command is used to attach molecule input files to a Batch calculation you are setting up. You must create the batch calculation (Batch Create) first, before you can attach molecule files to it. And you must attach molecule input files to a batch calculation before you can send it for execution (Run, below).
You can attach more than one molecular input file to the same batch, by repeatedly issuing the molecule create command with additional files.
-b/--batch <batch_name>
The name of an existing batch you have created. (To check what batch calculations you have created but not yet submitted, use the command “sbb batch list” and focus on those with “Staged” status. If the batch_name has embedded spaces, you must surround it with double quotes.
-f/--file <molecule_filename_and_path>
The name and path for the input molecular file.
-t/--type <sdf/smi/json>
The type of molecular input being supplied. Available options are sdf/smi/json. By default, the type is inferred from the filename extension (.sdf or .smi). If the user has specified –no_generate_conformers for this batch, then only SDF input is allowed, and an attempt to specify SMILES input will result in an error.
When a list of molecules is uploaded, it is parsed for errors, and molecules that fail that parsing will annotated with the problem detected. (Error status can be viewed using Batch List, below). When run from the command line, molecules that flag an error are automatically skipped when the job is submitted to run.
Molecule List¶
sbb molecule list -b/--batch <batch_name> [-o/--output-style <table/csv/html/json>]
Lists the molecules that have been uploaded to an existing batch with name batch_name. If there were errors detected for some of the uploaded molecules, these will annotated with the error status.
Molecule Delete¶
sbb molecule delete -b/--batch <batch_name> \
-i/--id <dataset_id>
Allows the user to delete specific molecules associated with the specified batch_name. The dataset_id for each molecule is in the first column in the table generated using molecule list:

You can specify multiple molecule dataset_id values separated by white spaces. For example:
sbb molecule delete -b CLI1 -i 174891 174892
Specifying the Config File (RC)¶
By default, a configuration file is automatically generated for the user during the installation process, and is found in the home directory: $HOME/.sbb_cli_rc
There is generally no reason for the user to examine or change this file and the default is sufficient. In rare cases, you may wish to use a non-default RC file (although typically only if instructed to do so by your sysadmin or QSimulate). In this case, any sbb command you issue can be post-pended by the option
-rc alternate_configuration_file
Which allows the user to specify an alternate RC file.
Example of Running a Calculation Using the CLI¶
In this example, we are going to assume you’ve set up the virtual environment, as described in the installation chapter, and called it QuantumFP. We’ll also assume you have followed the suggestion in that chapter, and created an alias called “quantumfp_cli” in your .bashrc file to activate the virtual environment. See the installation chapter for more details.
First, log into your Linux account. When you log in, your .bashrc file should automatically get parsed, setting up your virtual environment alias.
Now, activate the virtual environment where you have installed the CLI:
(base) Prompt$ conda activate QuantumFP
(QuantumFP) Prompt$
When you activate the virtual environment, the name of the environment will appear between parentheses at the beginning of your prompt. Once activated, the virtual environment stays activated for the remainder of your login session (or until you deactivate the session). The string shown for “Prompt” will depend on your account name and how your Linux machine was set up.
Let’s create a SMILES format input file that can be used with the CLI. (If you have your own SMILES or SDF input file already, you can use that).
Use your favorite file editor (emacs/vi/vim/etc) to create a file named “ThreeSmilesTest.smi”and insert the following three lines.
CC(=O)Oc1ccccc1C(=O)O ZINC000000000053
CC[C@H](C(=O)O)C1(O)CCCCC1 ZINC000000000194
CC(=O)Nc1ccc(C(=O)O)cc1 ZINC000000000226
Save this file. You’ll now have a file named ThreeSmilesTest.smi that has three small molecules taken from the ZINC screening database.
Log into your QSimulate CLI account.
(QuantumFP) Prompt$ sbb session set-url URL_TO_ACCESS_QSIMULATE
(QuantumFP) Prompt$ sbb session login --user YOUR_USER_NAME
Replace URL_TO_ACCESS_QSIMULATE with the URL provided by your system administrator or QSimulate. Replace YOUR_USER_NAME with the login name (or email) you use with the QSimulate account. You’ll be prompted to provide your password. If you are successful, you will see “Login successful.” on your screen.
Note that a login expires after 15 minutes of inactivity, and you’ll need to login again if that happens (using the same command).
At this point, you’ll want to create a new Batch calculation. You should first look at the names of any Batch calculations you have already run from this account, because you’ll need to assign a name, and it has to be unique. To look at the list of calculations you have already run, issue the command:
(QuantumFP) Prompt$ sbb batch list
This will show a table of all Batch calculations you have created, for example:
+------------------------------+--------+----------+
| Batch Name | smiles | status |
+------------------------------+--------+----------+
| First Run | 48 | complete |
| Run Number 2 | 149 | complete |
+------------------------------+--------+----------+
This table will include all Batch calculations you have created, whether from the CLI or the GUI. When creating a new Batch calculation, you need to choose a name not already in the table. (The CLI will let you know if you try to make a new Batch with the same name).
To create a new batch calculation use the command:
(QuantumFP) Prompt$ sbb batch create -b my_new_batch --generate_conformers -t basic
This command will create a new Batch calculation that you can work on, named “my_new_batch”. It indicates that conformers will be generated for each input molecule (required when using SMILES input), and it indicates that we will be calculating the “basic” set of descriptors. All other values are left at their defaults.
Next, we need to attach to this Batch calculation the list of molecules we want to use for input. To do this, we use the following command:
(QuantumFP) Prompt$ sbb molecule create -b my_new_batch --generate_conformers \
-f ./ThreeSmilesTest.smi
This command attaches the file ThreeSmilesTest.smi, which you created earlier, to the batch calculation my_new_batch. The command, as written, assumes the ThreeSmilesTest.smi file is in the directory you’re working from. If it isn’t, just add the correct path before the name of the file in the command.
Finally, we need to run the Batch calculation we’ve just set up:
(QuantumFP) Prompt$ sbb batch run -b my_new_batch
This will submit the Batch job to the CLI servers for execution.
Note that the CLI also offers the ability to combine the above three commands (batch create/molecule create/batch run) into a single command, create-and-run. This is often more convenient to use and offers all the run options available when issuing the commands separately. You could replace the above three commands with this single command:
(QuantumFP) Prompt$ sbb batch create-and-run -b my_new_batch --generate_conformers \
-t basic -f /ThreeSmilesTest.smi
When issuing commands from the command line, whether you issue the three commands separately, or use the create-and-run command from the command line is a matter of preference. But it is important to note that if you plan on running these commands from a script or unattended pipeline, it is strongly recommended you use only the create-and-run command. The reason for this is that there can be a lag in the molecule attachment step (depending on the size of the input molecules file), and if you attempt to issue the separate run command before the attachment is complete, the job will not submit. The create-and-run command will ensure that the actual run command waits for the molecule attachment to complete.
Using the “sbb batch list” command, you can track the progress of the job until it completes. Eventually, you’ll see the table for batch list look something like:
+------------------------------+--------+----------+
| Batch Name | smiles | status |
+------------------------------+--------+----------+
| First Run | 48 | complete |
| Run Number 2 | 149 | complete |
| my_new_batch | 3 | complete |
+------------------------------+--------+----------+
Once your job is complete, you can download the results. The following command will download a results table in CSV format:
(QuantumFP) Prompt$ sbb batch results -b my_new_batch -o csv > my_new_batch.csv
The results table will be in the file my_new_batch.csv. You can view this file using a text editor, or import it into a spreadsheet program like Excel.
For clarity, a summary of a simple CLI workflow is shown in the figure below.

The Molecular Fingerprints Command Line Interface (CLI): Installation¶
Installation Overview¶
Before you can run the QSimulate QuantumFP CLI, you need to install some infrastructure. This chapter describes the installation process. This is a straightforward process, only needs to be done once, and does not generally require administrative privileges.
The CLI will run from your local Linux host. Any standard modern Linux host can be used, including both dedicated Linux machines, as well as the “Linux Subsystem” that is supported in modern versions of Windows (versions 10 and 11). The CLI can also be installed and run from the Unix shell available as part of MacOS.
Installation requires that you download the “qysim” program package from QSimulate. You will download the package in whl (“wheel”) format, which can be installed using Python/Pip with a single command.
Before you install the qysim package, you will need to ensure you have the proper software infrastructure installed on your Linux installation. This is best carried out in a virtual environment, which serves multiple purposes:
This will allow you to install the necessary software without the need for “Administrator” privileges
This will ensure that the correct version of Python required to run the software is installed
This will isolate all the Python-installed software in a virtual container that cannot affect other software already installed on your computer
The implementation description that follows is for a Bash shell environment, which is the default shell for most Linux and Unix distributions. If you happen to use a different shell (e.g. tcsh, csh, etc.) you may need to modify the syntax of some of these commands, but the actual steps won’t change.
The process of installation is described in four parts:
Installation of miniconda3 (only performed one time; not necessary if already installed on your machine)
Creation of the QSimulate QuantumFP virtual environment
Installation of other software in the virtual environment that is required to run our software
Installation of the QSimulate software
Installing the virtual Environment (Miniconda)¶
If miniconda is already installed on your system, skip this section.
We are going to set up the CLI access within the Miniconda environment management system. There are alternatives to Miniconda (e.g. Venv and PyDev), but Miniconda has advantages, particularly with respect to ensuring you don’t wind up with mixed (and conflicting) versions of Python on your system. Miniconda is an efficient reduced-size-and-scope version of the venerable Anaconda environment manager, and has all the features we need. You only need to install Miniconda once. If you have already installed Miniconda on your system, you won’t need to install it again now.
If you are an advanced user and prefer a different approach to the virtual environment, that’s fine–but you’ll need to modify the commands described below to reflect the approach you use, and you’ll be responsible for ensuring an appropriate version of Python (3.8 or higher) is installed and that it doesn’t conflict with other installed software. Unless you are an advanced user, we strongly recommend you use miniconda as described.
To install Miniconda3 run the following commands. This will create a subdirectory named miniconda3 in your home directory that will contain both the miniconda program, and, subsequently, any Python packages you install while in the virtual environment.
These installation instructions assume you are using the Bash shell.
1) Log into your Linux environment.
2) cd $HOME
3) mkdir -p ~/miniconda3
4) wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh \
-O ~/miniconda3/miniconda.sh
5) cd miniconda3
6) chmod ugo+rwx miniconda.sh
7) bash ~/miniconda3/miniconda.sh -b -u -p ~/miniconda3
8) rm -rf ~/miniconda3/miniconda.sh
9) ~/miniconda3/bin/conda init bash
10) cd $HOME
11) source .bashrc
Upon sourcing your .bashrc, you will find that your default Prompt: is replaced by “(base) Prompt:”, which reflects the fact that Miniconda is now installed and working on your machine. (base) Prompt: means that while Miniconda is running, you have not yet entered any named virtual environment. You can install software in the (base) environment, but it is generally considered bad practice and not recommended.
Creating the QSimulate QuantumFP virtual environment¶
Now that we have installed Miniconda, we can create virtual environments within Miniconda. Each virtual environment (VE) is an independent branch of your installed operating system. You can activate (enter) and deactivate (exit) a VE at any time. When you activate the VE, you can access all the Python packages you installed in that VE, but you won’t Python packages you may have installed in different VEs. As a result, Python-related software you install in a VE can’t pollute your system or break dependencies that are assumed for other packages installed on your computer outside the VE. And if you make a mistake or don’t want to use a VE anymore, you can easily delete that VE–and all the Python packages installed to that VE–without affecting anything else on your computer.
For QuantumFP, we’ll create a VE named QuantumFP. In this VE, we’ll install a suitable version of Python and additional tools (Pip, Git, etc.) that are required for QuantumFP to install and run.
With these commands, we are creating a VE named QuantumFP. But you could use a different name, if desired. The conda create command creates the VE with a specific version of Python (3.8) that is suitable for our purposes. The conda activate command enters us into the created VE.
1) conda create --name QuantumFP python=3.8 -y
2) conda activate QuantumFP
Installing software in the QuantumFP virtual environment required to install/run QuantumFP¶
Before we install the actual QuantumFP package, we need to install some software that will be used during the installation process. The following commands take care of that installation. Be sure that you issued the conda activate QuantumFP command above, so that you are installing into the appropriate VE.
1) pip install -U setuptools setuptools_scm wheel
Installing QuantumFP (QYSIM)¶
1) Download the QYSIM package using the link Qsimulate provides.
(This will be a .whl ["wheel"] Python installer package).
We will call this QYSIM.whl here,
but the actual name may vary and you should substitute that name below.)
2) (While in the QuantumFP VE) cd to the directory where you downloaded QYSIM.whl
3) pip install QYSIM.whl
4) hash -r
Using QuantumFP on subsequent logins¶
When you log out of your Linux session, all virtual environments are automatically closed. When you log in again, you will need to active (reopen) the VE you want to use. If you followed the installation instructions above, your VE for QuantumFP is, itself, named QuantumFP. To activate the VE to use it you need to issue the command:
conda activate QuantumFP
If everything is working properly, after issuing the above command, you should find your default prompt replaced as below
(base) Prompt:
(base) Prompt: conda activate QuantumFP
(QuantumFP) Prompt:
Testing QuantumFP CLI¶
At this point, your CLI installation under the virtual environment should be working. To test it, try the following four commands (ensuring, first, that your QuantumFP virtual environment is active):
(QuantumFP) Prompt: sbb session set-url --url YOUR_QSIMULATE_ACCESS_URL
(QuantumFP) Prompt: sbb session login --user YOUR_QSIMULATE_EMAIL_LOGIN
(QuantumFP) Prompt: sbb batch list
(QuantumFP) Prompt: sbb session logout
Where YOUR_QSIMULATE_ACCESS_URL is replaced by the URL you were provided by QSimulate or your admin, and YOUR_QSIMULATE_EMAIL_LOGIN is replaced by the email address associated with your account. If your installation is properly set up, the first two commands above should log you into the platform (you should see “Login successful” after you specify your password following the login command). The batch list command should execute without an error. And the logout command will disconnect you from the session, and you should see “Logged out.” appear at your terminal.
Exiting the Virtual Environment¶
If you wish to exit the virtual environment, issue the command “conda deactivate”. This will drop you back to the (base) level of Miniconda.
(QuantumFP) Prompt: conda deactivate
(base) Prompt:
Removing the virtual environment and QuantumFP installation¶
One of the great advantages of having installed QuantumFP in a virtual environment is that that makes it trivially easy to remove the installation. For example, if you wish to do a clean update installation, you can just remove the previous virtual environment, recreate the virtual environment, and then install the new version. (You can also have multiple virtual environments, if you wish to install the new update without removing the previous version).
If you have installed a number of different packages in the Miniconda environment manager, and only want to delete the QuantumFP installation (while keeping everything else as-is), you merely have to exit the VE using the command “deactivate” and then remove the VE, as follows:
(QuantumFP) Prompt: deactivate
(base) Prompt: conda env remove --name QuantumFP
(base) rm $HOME/.sbb_cli_rc
The last of these commands deletes a configuration file that QuantumFP will have created in your home directory.
To see a list of all virtual environments installed in Miniconda, you can use the “env list” command:
(base) Prompt: conda env list